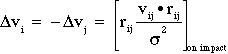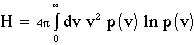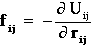
Pair force:
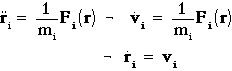
Newton's equation of motion:
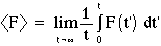
6N coupled first order differential equations.
Method: the 6N coupled equations are supplemented with 6N initial conditions (for example, 3N coordinates at one time and 3N coordinates at a later time, or 3N coordinates and 3N velocities at a common time) and solved numerically. Common algorithms are the Verlet algorithms or Predictor-Corrector algorithms (e.g. Euler-type predictor with trapezoid corrector).
Properties are then computed from samples along the trajectory:
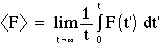
for simple thermodynamic averages, like <U>, <V> etc. The total internal energy is the sum of the potential and kinetic contributions:
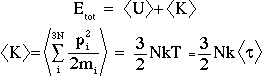
<tau> is a "configuration temperature".
Pressure (if volume is allowed to vary during the simulation):
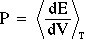
Pressure (if volume is held constant during the simulation):
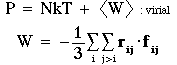
Note that for small molecules it is conventional to associate pressure with intermolecular forces, where f_ij is the force between molecules i and j. For polymers, this distinction becomes vague, and it is important to distinguish between the "molecular virial" and the "atomic virial".
Combinatorial properties are implicit in the MD ensemble, but may be obtained relative to a known thermodynamic state by the use of thermodynamic integration:

Other quantities may be found from fluctuations in quantities like energy, volume, etc. For more detail, refer to Allen and Tildesley, Chapter 2.
Molecular Dynamics, by nature, samples phase space using equations of motion which are energy-conserving, that is, constant NVE. Methods have been proposed to "rescale" the equations of motion so that other ensembles are samples (e.g. for NVT, see S. Nose, J. Chem. Phys., 81, 571, 1984; for NPT see M. Parinello and A. Rahman, J. Appl. Phys., 52, 7182, 1981.)
However, one of the real advantages of MD is that it allows estimation of dynamic quantities (i.e. transport coefficients) in NVE.
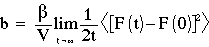
Green Kubo Relation (ca. 1955)
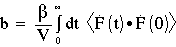
In this manner, any molecular property F can be related to the corresponding transport coefficient b:
coefficient of self diffusion
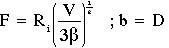
coefficient of thermal conductivity
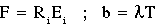
static electrical conductivity
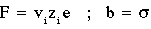
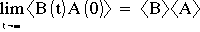
It is common to subtract the limiting value from the correlation function and then to normalize this function to get a function which decays from 1 to 0. A related function is the Fluctuation Correlation:
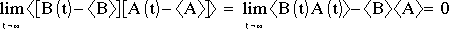
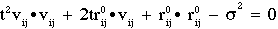
Upon impact, collision is assumed elastic, so: