Reprinted with permission, IEEE Computer, Vol. 26, No. 7, July 1993, pp. 18-41.
Recall that the Tyler error occurred when the operator made an entry indicating the mode/energy, went to the command line, then moved the cursor up to change the mode/energy, and returned to the command line all within 8 seconds. Since the magnet setting takes about 8 seconds and Magnet does not recognize edits after the first execution of Ptime, the editing had been completed by the return to Datent, which never detected that it had occurred. Part of the problem was fixed after the accident by clearing the bending-magnet variable at the end of Magnet (after all the magnets have been set) instead of at the end of Ptime.
But this was not the only problem. Upon exit from the Magnet subroutine, the data-entry subroutine (Datent) checks the data-entry completion variable. If it indicates that data entry is complete, Datent sets Tphase to 3 and Datent is not entered again. If it is not set, Datent leaves Tphase unchanged, which means it will eventually be rescheduled. But the data-entry completion variable only indicates that the cursor has been down to the command line, not that it is still there. A potential race condition is set up. To fix this, AECL introduced another shared variable controlled by the keyboard handler task that indicates the cursor is not positioned on the command line. If this variable is set, then prescription entry is still in progress and the value of Tphase is left unchanged.
Government and user response. The FDA does not approve each new medical device on the market: All medical devices go through a classification process that determines the level of FDA approval necessary. Medical accelerators follow a procedure called pre-market notification before commercial distribution. In this process, the firm must establish that the product is substantially equivalent in safety and effectiveness to a product already on the market. If that cannot be done to the FDA's satisfaction, a pre-market approval is required. For the Therac-25, the FDA required only a pre-market notification.
The agency is basically reactive to problems and requires manufacturers to report serious ones. Once a problem is identified in a radiation-emitting product, the FDA must approve the manufacturer's corrective action plan (CAP).
The first reports of the Tyler accidents came to the FDA from the state of Texas health department, and this triggered FDA action. The FDA investigation was well under way when AECL produced a medical device report to discuss the details of the radiation overexposures at Tyler. The FDA declared the Therac-25 defective under the Radiation Control for Health and Safety Act and ordered the firm to notify all purchasers, investigate the problem, determine a solution, and submit a corrective action plan for FDA approval.
The final CAP consisted of more than 20 changes to the system hardware and software, plus modifications to the system documentation and manuals. Some of these changes were unrelated to the specific accidents, but were improvements to the general machine safety. The full implementation of the CAP, including an extensive safety analysis, was not complete until more than two years after the Tyler accidents.
AECL made its accident report to the FDA on April 15, 1986. On that same date, AECL sent a letter to each Therac user recommending a temporary "fix" to the machine that would allow continued clinical use. The letter (shown in its complete form) read as follows:
SUBJECT: CHANGE IN OPERATING PROCEDURES FOR THE THERAC 25 LINEAR ACCELERATOROn May 2, 1986, the FDA declared the Therac defective, demanded a CAP, and required renotification of all the Therac customers. In the letter from the FDA to AECL, the director of compliance, Center for Devices and Radiological Health, wrote
Effective immediately, and until further notice, the key used for moving the cursor back through the prescription sequence (i.e., cursor "UP" inscribed with an upward pointing arrow) must not be used for editing or any other purpose.
To avoid accidental use of this key, the key cap must be removed and the switch contacts fixed in the open position with electrical tape or other insulating material. For assistance with the latter you should contact your local AECL service representative.
Disabling this key means that if any prescription data entered is incorrect then [an] "R" reset command must be used and the whole prescription reentered.
For those users of the Multiport option, it also means that editing of dose rate, dose, and time will not be possible between ports.
We have reviewed Mr. Downs' April 15 letter to purchasers and have concluded that it does not satisfy the requirements for notification to purchasers of a defect in an electronic product. Specifically, it does not describe the defect nor the hazards associated with it. The letter does not provide any reason for disabling the cursor key and the tone is not commensurate with the urgency for doing so. In fact, the letter implies the inconvenience to operators outweighs the need to disable the key. We request that you immediately renotify purchasers.
AECL promptly made a new notice to users and also requested an extension to produce a CAP. The FDA granted this request.
About this time, the Therac-25 users created a user group and held their first meeting at the annual conference of the American Association of Physicists in Medicine. At the meeting, users discussed the Tyler accident and heard an AECL representative present the company's plans for responding to it. AECL promised to send a letter to all users detailing the CAP.
Several users described additional hardware safety features that they had added to their own machines to provide additional protection. An interlock (that checked gun current values), which the Vancouver clinic had previously added to its Therac-25, was labeled as redundant by AECL. The users disagreed. There were further discussions of poor design and other problems that caused 10- to 30-percent underdosing in both modes.
The meeting notes said
. . . there was a general complaint by all users present about the lack of information propagation. The users were not happy about receiving incomplete information. The AECL representative countered by stating that AECL does not wish to spread rumors and that AECL has no policy to "keep things quiet." The consensus among the users was that an improvement was necessary.
After the first user group meeting, there were two user group newsletters. The first, dated fall 1986, contained letters from Still, the Kennestone physicist, who complained about what he considered to be eight major problems he had experienced with the Therac-25. These problems included poor screen-refresh subroutines that left trash and erroneous information on the operator console, and some tape-loading problems upon start-up, which he discovered involved the use of "phantom tables" to trigger the interlock system in the event of a load failure instead of using a check sum. He asked the question, "Is programming safety relying too much on the software interlock routines?" The second user group newsletter, in December 1986, further discussed the implications of the "phantom table" parameterization.
AECL produced the first CAP on June 13, 1986. It contained six items:
(1) Fix the software to eliminate the specific behavior leading to the Tyler problem.
(2) Modify the software sample-and-hold circuits to detect one pulse above a nonadjustable threshold. The software sample-and-hold circuit monitors the magnitude of each pulse from the ion chambers in the beam. Previously, three consecutive high readings were required to shut off the high-voltage circuits, which resulted in a shutdown time of 300 ms. The software modification results in a reading after each pulse, and a shutdown after a single high reading.
(3) Make Malfunctions 1 through 64 result in treatment suspend rather than pause.
(4) Add a new circuit, which only administrative staff can reset, to shut down the modulator if the sample-and-hold circuits detect a high pulse. This is functionally equivalent to the circuit described in item 2. However, a new circuit board is added that monitors the five sample-and-hold circuits. The new circuit detects ion-chamber signals above a fixed threshold and inhibits the trigger to the modulator after detecting a high pulse. This shuts down the beam independently of the software.
(5) Modify the software to limit editing keys to cursor up, backspace, and return.
(6) Modify the manuals to reflect the changes.
FDA internal memos describe their immediate concerns regarding the CAP. One memo suggests adding an independent circuit that "detects and shuts down the system when inappropriate outputs are detected," warnings about when ion chambers are saturated, and under-standable system error messages. Another memo questions "whether all possible hardware options have been investigated by the manufacturer to prevent any future inadvertent high exposure."
On July 23 the FDA officially responded to AECL's CAP submission. They conceptually agreed with the plan's direction but complained about the lack of specific information necessary to evaluate the plan, especially with regard to the software. The FDA requested a detailed description of the software- development procedures and documentation, along with a revised CAP to include revised the software setup table, and the software interlock interactions. The FDA also made a very detailed request for a documented test plan, and detailed descriptions of the revised edit modes, the changes made to the software setup table, and the software interlock interactions. The FDA also made a very detailed request for a documented test plan.
AECL responded on September 26 with several documents describing the software and its modifications but no test plan. They explained how the Therac-25 software evolved from the Therac-6 software and stated that "no single test plan and report exists for the software since both hardware and software were tested and exercised separately and together over many years." AECL concluded that the current CAP improved "machine safety by many orders of magnitude and virtually eliminates the possibility of lethal doses as delivered in the Tyler incident."
An FDA internal memo dated October 20 commented on these AECL submissions, raising several concerns:
Unfortunately, the AECL response also seems to point out an apparent lack of documentation on software specifications and a software test plan.. . . concerns include the question of previous knowledge of problems by AECL, the apparent paucity of software QA [quality assurance] at the manufacturing facility, and possible warnings and information dissemination to others of the generic type problems.
. . . As mentioned in my first review, there is some confusion on whether the manufacturer should have been aware of the software problems prior to the [accidental radiation overdoses] in Texas. AECL had received official notification of a lawsuit in November 1985 from a patient claiming accidental over-exposure from a Therac-25 in Marietta, Georgia. . . If knowledge of these software deficiencies were known beforehand, what would be the FDA's posture in this case?
. . . The materials submitted by the manufacturer have not been in sufficient detail and clarity to ensure an adequate software QA program currently exists. For example, a response has not been provided with respect to the software part of the CAP to the CDRH [FDA Center for Devices and Radiological Health] request for documentation on the revised requirements and specifications for the new software. In addition, an analysis has not been provided, as requested, on the interaction with other portions of the software to demonstrate the corrected software does not adversely affect other software functions.
The July 23 letter from the CDRH requested a documented test plan includ-ing several specific pieces of information identified in the letter. This request has been ignored up to this point by the manufacturer. Considering the ramifi-cations of the current software problem, changes in software QA attitudes are needed at AECL.
On October 30, the FDA responded to AECL's additional submissions, complaining about the lack of a detailed description of the accident and of sufficient detail in flow diagrams. Many specific questions addressed the vagueness of the AECL response and made it clear that additional CAP work must precede approval.
AECL, in response, created CAP Revision 1 on November 12. This CAP contained 12 new items under "software modifications," all (except for one cosmetic change) designed to eliminate potentially unsafe behavior. The submission also contained other relevant documents including a test plan.
The FDA responded to CAP Revision 1 on December 11. The FDA explained that the software modifications appeared to correct the specific deficiencies discovered as a result of the Tyler accidents. They agreed that the major items listed in CAP Revision 1 would improve the Therac's operation. However, the FDA required AECL to attend to several further system problems before CAP approval. AECL had proposed to retain treatment pause for some dose-rate and beam-tilt malfunctions. Since these are dosimetry system problems, the FDA considered them safety interlocks and believed treatment must be suspended for these malfunctions.
AECL also planned to retain the malfunction codes, but the FDA required better warnings for the operators. Furthermore, AECL had not planned on any quality assurance testing to ensure exact copying of software, but the FDA insisted on it. The FDA further requested assurances that rigorous testing would become a standard part of AECL's software-modification procedures:
We also expressed our concern that you did not intend to perform the protocol to future modifications to software. We believe that the rigorous testing must be performed each time a modification is made in order to ensure the modification does not adversely affect the safety of the system.
AECL was also asked to draw up an installation test plan to ensure both hardware and software changes perform as designed when installed.
AECL submitted CAP Revision 2 and supporting documentation on December 22, 1986. They changed the CAP to have dose malfunctions suspend treatment and included a plan for meaningful error messages and highlighted dose error messages. They also expanded diagrams of software modifications and expanded the test plan to cover hardware and software.
On January 26, 1987, AECL sent the FDA their "Component and Instal-lation Test Plan" and explained that their delays were due to the investigation of a new accident on January 17 at Yakima.
Yakima Valley Memorial Hospital, 1987. On Saturday, January 17, 1987, the second patient of the day was to be treated at the Yakima Valley Memorial Hospital for a carcinoma. This patient was to receive two film-verification exposures of 4 and 3 rads, plus a 79-rad photon treatment (for a total exposure of 86 rads).
Film was placed under the patient and 4 rads was administered with the collimator jaws opened to 22 x 18 cm. After the machine paused, the collimator jaws opened to 35 x 35 cm automatically, and the second exposure of 3 rads was administered. The machine paused again.
The operator entered the treatment room to remove the film and verify the patient's precise position. He used the hand control in the treatment room to rotate the turntable to the field-light position, a feature that let him check the machine's alignment with respect to the patient's body to verify proper beam position. The operator then either pressed the set button on the hand control or left the room and typed a set command at the console to return the turntable to the proper position for treatment; there is some confusion as to exactly what transpired. When he left the room, he forgot to remove the film from underneath the patient. The console displayed "beam ready," and the operator hit the "B" key to turn the beam on.
The beam came on but the console displayed no dose or dose rate. After 5 or 6 seconds, the unit shut down with a pause and displayed a message. The message "may have disappeared quickly"; the operator was unclear on this point. However, since the machine merely paused, he was able to push the "P" key to proceed with treatment.
The machine paused again, this time displaying "flatness" on the reason line. The operator heard the patient say something over the intercom, but couldn't understand him. He went into the room to speak with the patient, who reported "feeling a burning sensation" in the chest. The console displayed only the total dose of the two film exposures (7 rads) and nothing more.
Later in the day, the patient developed a skin burn over the entire treatment area. Four days later, the redness took on the striped pattern matching the slots in the blocking tray. The striped pattern was similar to the burn a year earlier at this hospital that had been attributed to "cause unknown."
AECL began an investigation, and users were told to confirm the turntable position visually before turning on the beam. All tests run by the AECL engineers indicated that the machine was working perfectly. From the information gathered to that point, it was suspected that the electron beam had come on when the turntable was in the field-light position. But the investigators could not reproduce the fault condition that produced the overdose.
On the following Thursday, AECL sent an engineer from Ottawa to investigate. The hospital physicist had, in the meantime, run some tests with film. He placed a film in the Therac's beam and ran two exposures of X-ray parameters with the turntable in field-light position. The film appeared to match the film that was left (by mistake) under the patient during the accident.
After a week of checking the hardware, AECL determined that the "incorrect machine operation was probably not caused by hardware alone." After checking the software, AECL discovered a flaw (described in the next section) that could explain the erroneous behavior. The coding problems explaining this accident differ from those associated with the Tyler accidents.
AECL's preliminary dose measurements indicated that the dose delivered under these conditions - that is, when the turntable was in the field-light position - was on the order of 4,000 to 5,000 rads. After two attempts, the patient could have received 8,000 to 10,000 instead of the 86 rads prescribed. AECL again called users on January 26 (nine days after the accident) and gave them detailed instructions on how to avoid this problem. In an FDA internal report on the accident, an AECL quality assurance manager investigating the problem is quoted as saying that the software and hardware changes to be retrofitted following the Tyler accident nine months earlier (but which had not yet been installed) would have prevented the Yakima accident.
The patient died in April from complications related to the overdose. He had been suffering from a terminal form of cancer prior to the radiation overdose, but survivors initiated lawsuits alleging that he died sooner than he would have and endured unnecessary pain and suffering due to the overdose. The suit was settled out of court.
The Yakima software problem. The software problem for the second Yakima accident is fairly well established and different from that implicated in the Tyler accidents. There is no way to determine what particular software design errors were related to the Kennestone, Hamilton, and first Yakima accidents. Given the unsafe programming practices exhibited in the code, it is possible that unknown race conditions or errors could have been responsible. There is speculation, however, that the Hamilton accident was the same as this second Yakima overdose. In a report of a conference call on January 26, 1987, between the AECL quality assurance manager and Ed Miller of the FDA discussing the Yakima accident, Miller notes
This situation probably occurred in the Hamilton, Ontario, accident a couple of years ago. It was not discovered at that time and the cause was attributed to intermittent interlock failure. The subsequent recall of the multiple microswitch logic network did not really solve the problem.
The second Yakima accident was again attributed to a type of race condition in the software - this one allowed the device to be activated in an error setting (a "failure" of a software interlock). The Tyler accidents were related to problems in the data-entry routines that allowed the code to proceed to Set-Up Test before the full prescription had been entered and acted upon. The Yakima accident involves problems encountered later in the logic after the treatment monitor Treat reaches Set-Up Test.
The Therac-25's field-light feature permits very precise positioning of the patient for treatment. The operator can control the Therac-25 right at the treatment site using a small hand control offering certain limited functions for patient setup, including setting gantry, collimator, and table motions.
Normally, the operator enters all the prescription data at the console (outside the treatment room) before the final setup of all machine parameters is completed in the treatment room. This gives rise to an "unverified" condition at the console. The operator then completes the patient setup in the treatment room, and all relevant parameters now "verify." The console displays the message "Press set button" while the turntable is in the field-light position. The operator now presses the set button on the hand control or types "set" at the console. That should set the collimator to the proper position for treatment.
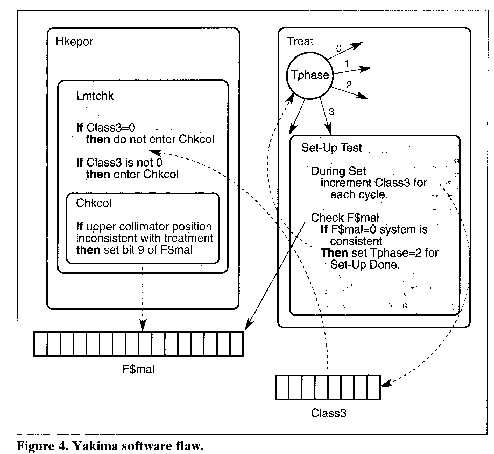
In the software, after the prescription is entered and verified by the Datent routine, the control variable Tphase is changed so that the Set-Up Test routine is entered (see Figure 4 Yakima software flaw). Every pass through the Set-Up Test routine increments the upper collimator position check, a shared variable called Class3. If Class3 is nonzero, there is an inconsistency and treatment should not proceed. A zero value for Class3 indicates that the relevant parameters are consistent with treatment, and the beam is not inhibited.
After setting the Class3 variable, Set-Up Test next checks for any malfunctions in the system by checking another shared variable (set by a routine that actually handles the interlock checking) called F$mal to see if it has a nonzero value. A nonzero value in F$mal indicates that the machine is not ready for treatment, and the Set-Up Test subroutine is rescheduled. When F$mal is zero (indicating that everything is ready for treatment), the Set-Up Test subroutine sets the Tphase variable equal to 2, which results in next scheduling the Set-Up Done subroutine, and the treatment is allowed to continue.
The actual interlock checking is performed by a concurrent Housekeeper task (Hkeper). The upper collimator position check is performed by a subroutine of Hkeper called Lmtchk (analog/digital limit checking). Lmtchk first checks the Class3 variable. If Class3 contains a nonzero value, Lmtchk calls the Check Collimator (Chkcol) subroutine. If Class3 contains zero, Chkcol is bypassed and the upper collimator position check is not performed. The Chkcol subroutine sets or resets bit 9 of the F$mal shared variable, depending on the position of the upper collimator (which in turn is checked by the Set-Up Test subroutine of Datent so it can decide whether to reschedule itself or proceed to Set-Up Done).
During machine setup, Set-Up Test will be executed several hundred times since it reschedules itself waiting for other events to occur. In the code, the Class3 variable is incremented by one in each pass through Set-Up Test. Since the Class3 variable is 1 byte, it can only contain a maximum value of 255 decimal. Thus, on every 256th pass through the Set-Up Test code, the variable overflows and has a zero value. That means that on every 256th pass through Set-Up Test, the upper collimator will not be checked and an upper collimator fault will not be detected.
The overexposure occurred when the operator hit the "set" button at the precise moment that Class3 rolled over to zero. Thus Chkcol was not executed, and F$mal was not set to indicate the upper collimator was still in field-light position. The software turned on the full 25 MeV without the target in place and without scanning. A highly concentrated electron beam resulted, which was scattered and deflected by the stainless steel mirror that was in the path.
AECL described the technical "fix" implemented for this software flaw as simple: The program is changed so that the Class3 variable is set to some fixed nonzero value each time through Set-Up Test instead of being incremented.
{kind=link}