|
|
||
 |
 |
 |
|
|
|
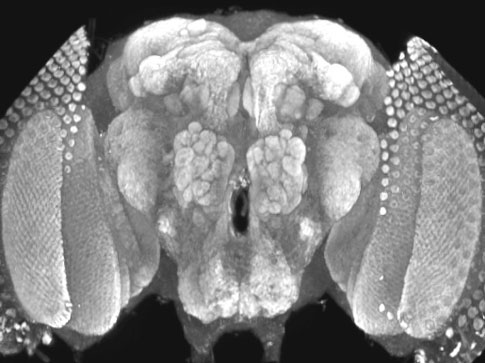
Huntington’s Disease
Regarding the long-term goals for HD research in the lab, we have set out a path to address two essential problems that must be solved in order to eventually develop therapeutic strategies that might alter the course of HD. First, we must establish genetic models of the disease that mimic at least parts of the in vivo pathogenesis in HD patients that will allow us to identify genetic or pharmacological suppressors of Httex-mediated neuronal dysfunction. With a long-term vision of finding pathways or reagents that can serve as potential therapeutic targets in HD, we will need to develop an understanding of how neurodegeneration and polyQ expansion are linked. The second important experimental advance we need to consider in order to translate inhibitors of Httex-mediated neuronal dysfunction into therapeutic targets is the generation of a model that will allow testing for the effects of inhibitors on the normal function of endogenous Htt. As a cure for HD may ultimately involve direct manipulation of Htt activity, we must understand the consequences of manipulating Htt function. Any inhibitor that prevents Httex pathogenesis or aggregation, but also disrupts normal Htt function, would not likely be an appropriate therapeutic target. In order to assay wildtype Htt function, we need to develop assays that provide an appropriate readout of the protein’s activity. Therefore, we are mutating the Htt locus in Drosophila and characterizing the resulting phenotypes. In addition, we are attempting to rescue Drosophila Htt mutants using human htt cDNAs encoding normal and pathogenic polyQ tracts. In this manner, we can then test whether suppressors or inhibitors of Httex pathogenesis prevent the ability of the human transgenes to rescue. If they allow rescue, we can identify pathways or inhibitors that block Httex-mediated neurodegeneration, but that do not affect the essential requirement for Htt in viability. |
|
 Huntington's
disease (HD) is an autosomal dominant neurodegenerative disorder caused by
expansion of a polyglutamine (polyQ) tract in the huntingtin (Htt) protein that
results in intracellular aggregate formation and neurodegeneration. Pathways
leading from polyQ tract expansion to disease pathogenesis remain obscure. To
elucidate how polyQ expansion causes neuronal dysfunction, we have generated
Drosophila transgenic strains expressing human htt cDNAs encoding
pathogenic or nonpathogenic proteins. While expression of nonpathogenic Htt has
no discernible effect on behavior, lifespan or neuronal morphology, pan-neuronal
expression of Htt containing a pathogenic polyQ tract causes a progressive loss
of motor coordination, decreased lifespan and time-dependent formation of Htt
aggregates specifically in the cytoplasm and neurites. Htt aggregates
accumulate at synapses and along axonal tracts, resulting in disruptions of
axonal transport. In contrast, Drosophila expressing an expanded polyQ
tract alone, or an expanded polyQ tract in the context of the spinocerebellar
ataxia type 3 protein, display only nuclear aggregates and do not disrupt axonal
trafficking. Our findings indicate that non-nuclear events induced by
cytoplasmic huntingtin aggregation may play a central role in the progressive
neurodegeneration observed in Huntington’s disease.
Huntington's
disease (HD) is an autosomal dominant neurodegenerative disorder caused by
expansion of a polyglutamine (polyQ) tract in the huntingtin (Htt) protein that
results in intracellular aggregate formation and neurodegeneration. Pathways
leading from polyQ tract expansion to disease pathogenesis remain obscure. To
elucidate how polyQ expansion causes neuronal dysfunction, we have generated
Drosophila transgenic strains expressing human htt cDNAs encoding
pathogenic or nonpathogenic proteins. While expression of nonpathogenic Htt has
no discernible effect on behavior, lifespan or neuronal morphology, pan-neuronal
expression of Htt containing a pathogenic polyQ tract causes a progressive loss
of motor coordination, decreased lifespan and time-dependent formation of Htt
aggregates specifically in the cytoplasm and neurites. Htt aggregates
accumulate at synapses and along axonal tracts, resulting in disruptions of
axonal transport. In contrast, Drosophila expressing an expanded polyQ
tract alone, or an expanded polyQ tract in the context of the spinocerebellar
ataxia type 3 protein, display only nuclear aggregates and do not disrupt axonal
trafficking. Our findings indicate that non-nuclear events induced by
cytoplasmic huntingtin aggregation may play a central role in the progressive
neurodegeneration observed in Huntington’s disease.