
April 1998
Abstract
The possible trends for atmospheric carbon monoxide in the next 100 years have been illustrated using a coupled atmospheric chemistry and climate model driven by emissions predicted by a global economic development model. Various model runs with different assumptions regarding emissions or model parameters have been carried out to investigate the impacts of model and emission uncertainties on the predictions. We have found complicated interactions among emissions, atmospheric chemistry, and climate regarding the distributions and evolution of CO in the atmosphere. Based on the predicted emissions of methane and carbon monoxide, the model predicts an increasing trend of carbon monoxide in the next century with a tropospheric mole fraction of CO in 2100 double its present-day value.
Methane emissions are found to have the most important effect on the future atmospheric CO budget. High methane emissions cause significant depletion of tropospheric OH, increase of CO concentrations, and lengthening of lifetimes of many chemical species including CO and CH4. The global average atmospheric lifetime of CO is predicted in our reference model run to be about 0.6 month longer than its present value (~2 months). The predicted emissions of CO increase only slightly over the next century, so the impact of CO emissions on the predicted CO abundance appears to be less important than that of methane. Consequently, maintaining the emissions of CH4 at their current levels can prevent significant future changes in tropospheric chemistry, while similar controlling emissions of CO cannot achieve the same result.
This study also indicates that climate variations, especially those causing changes in H2O concentrations, can influence atmospheric trends of carbon monoxide. A two-way interaction between chemistry and climate regarding CO is evident. Specifically, the budget of atmospheric CO affects the destruction of methane and the production of CO2, ozone, and sulfate aerosols and thus affects climate, while the resultant changes in climate modify the budget of CO-CH4 in turn through their effects on H2O and temperature.
1. Introduction
In the troposphere, hydroxyl free radicals (OH) oxidize carbon monoxide (CO) to form hydroperoxy radicals (HO2) [Levy, 1971]:
CO + OH CO2 + H [R1]
H + O2 + M HO2 + M [R2]
As indicated by Crutzen and Zimmermann [1991], in a NOx-rich (here NOx = NO + NO2) environment, reactions following R1 and R2 produce ozone. Otherwise, reactions of ozone with hydroperoxy radicals destroy the ozone. Either reaction chain is a CO sink, producing carbon dioxide. The fast reaction R1 does not depend on temperature [DeMore et al., 1994]; it leads to a global-average CO lifetime of two months in the current atmosphere, and is the most significant OH-radical sink in the troposphere [e.g., Sze, 1977; Crutzen and Zimmermann, 1991]. Within the atmosphere, the major CO source is oxidation of methane by OH, producing formaldehyde (CH2O) and then carbon monoxide. This methane reaction, in combination with R1, consumes most OH radicals in the troposphere. The tropospheric abundances of CO and CH4 thus become important indices of the tropospheric oxidizing capacity, represented most specifically by the concentrations of ozone and hydroxyl free radicals.
Analyses on observational and ice-core data indicate that the tropospheric concentration of CH4 has increased rapidly since the start of the industrial era [Rasmussen and Khalil, 1984; Khalil and Rasmussen, 1987; Etheridge et al., 1992]. Current observations and model estimates suggest that CO concentrations have also increased greatly from pre-industrial levels [e.g., Thompson and Cicerone, 1986; Khalil and Rasmussen, 1990]. Increased anthropogenic emissions are believed to be responsible for the growing atmospheric concentrations of CO and CH4, and could possibly cause climate changes. Increased CO and CH4 emissions most likely also deplete hydroxyl radicals in the troposphere (e.g., Sze, 1971; Thompson and Cicerone, 1986; Crutzen and Zimmermann, 1991; Wang and Prinn, 1998a], affecting atmospheric chemistry. Recent observations have shown that CH4 and CO concentration increases slowed in the early 1990s [Novelli et al., 1994; Khalil and Rasmussen, 1995]. While no convincing explanation for this phenomenon has been advanced, possible reasons range from source reduction to changed hydroxyl-radical concentrations. Indeed, besides purely atmospheric chemical processes, climatic variations such as disturbances in temperature and H2O can also change the abundance of CO and CH4 [Klonecki and Levy, 1997; Wang and Prinn, 1998a]. Obviously, the evolution of atmospheric CO involves interactions among anthropogenic emissions, atmospheric chemistry, and climate.
In order to investigate these interactions and their influences on atmospheric composition and climate, we have developed a coupled chemistry and climate model to comprehensively describe climate dynamics and atmospheric chemical processes [Wang et al., 1998]. In this study, we employed this model to study the long-term (100-year) evolution of atmospheric CO and CH4. We first briefly describe the model and the designs of numerical experiments. The influence of anthropogenic emissions on atmospheric CO and climate are then discussed based on the results of these experiments. This is followed by a discussion of climate-chemistry interactions involving atmospheric CO, and conclusions.
Model and Numerical Experiment Design
The coupled atmospheric chemistry and climate model of Wang et al. [1998] is used to predict the future evolution of atmospheric carbon monoxide and to investigate the relative effects of emissions, atmospheric chemistry, and climate on carbon monoxide concentrations. The model is two-dimensional, with 24 latitude bands and nine pressure levels (two in the stratosphere, seven in the troposphere), using time steps of 20 minutes for climate dynamics and transport of chemicals, three hours for gaseous phase chemistry, and four hours for radiation. The chemistry model contains 25 chemical species, including carbon dioxide, nitrous oxide, chlorofluorocarbons, methane, carbon monoxide, ozone, NOx, HOx, sulfur dioxide, and sulfuric acid. Forty-one gas-phase and 12 heterogeneous reactions are included in the model. These chemical processes, along with emissions, dry and wet deposition, transport by winds, subgrid-scale mixing, and convection, serve as local sinks or sources of various chemical species in the atmosphere. The climate model provides wind speeds, air pressure, temperature, precipitation, convection parameters, and radiative fluxes at each model grid point and every time step for the chemistry model. The predicted concentrations of carbon dioxide, nitrous oxide, chlorofluorocarbons, methane, and sulfate aerosols from the chemical model are used in the climate model to calculate atmospheric radiative fluxes.
Emissions of chemicals include emissions from both natural and anthropogenic sources. Here we treat total annual natural emissions as constant with time. The annual natural emissions of CH4, N2O, CO, NO (excluding lightning), and sulfur are 130 Tg (CH4), 9.1 Tg (N), 158.6 Tg (C), 10 Tg (N), and 12.8 Tg (S), respectively. Lightning-produced NO is assumed to be 5 Tg (N). Anthropogenic emissions after 1985 are from predictions of the MIT Emission Prediction and Policy Analysis (EPPA) model [see Prinn et al., 1998, and Yang et al., 1996].
All numerical experiments in this study involved integrations from 1977 to 2100, allowing comparison of the earliest model predictions (1977-1995) with observational data. To illustrate a range of plausible CO predictions involving differing assumptions about emissions, chemistry, and climate, we conducted five experiments we chose in each case one of three assumptions regarding: (a) anthropogenic emissions, (b) the aerosol radiative forcing intensity and speed of oceanic uptake of heat and CO2, and (c) the climate model sensitivity to doubling CO2. Three labels, identifying the assumptions we made regarding (a), (b), and (c) are used to designate each experiment. The five runs are specifically labeled RRR, HRR, LRR, RHH, and RLL. "H" designates the high case, "R" the reference, and "L" the low case, with the choice of the label determined by the effect of the chosen assumptions on predicted surface temperature in 2100. For example, HRR is a run using a high emission prediction, reference settings for the aerosol radiative forcing intensity and speed of oceanic uptake, and reference settings for the climate model sensitivity. Three sets of anthropogenic emission predictions from the EPPA model for CO2, CH4, N2O, CO, NO, and SOx anthropogenic emissions are used. Combined with the assumed constant natural emissions, total predicted CO emissions in the high, reference, and low cases are 894, 864, and 837 Tg C/yr in 2100, respectively with all cases starting from 709 Tg C/yr in 1977 (Figure 1). The average increase rates are 0.19%/yr for the high-, 0.16%/yr for the reference-, and 0.14%/yr for the low-emission predictions. The discontinuities in emissions in Figure 1 in 1985 and 2050 are caused by the initiation of the EPPA predictions in 1985 and the assumed phase out of the anthropogenic deforestation source of CO in 2050. Predicted anthropogenic emissions of other species, especially methane, increased much faster than predicted emissions of CO [Wang and Prinn, 1998b]. For example, average growth rates of methane emissions in the high, reference, and low predictions are 0.65%/yr, 0.58%/yr, and 0.5%/yr, respectively. These differences are due to predicted growth in CH4 emissions from urban waste, agriculture, and mining (coal, gas) outstripping growth in the combustion-related (biomass, fossil fuel) sources of CO.
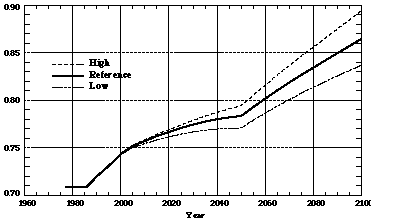
Figure 1. Predicted annual emissions of CO in high (H), reference (R), and low (L) cases in units of Pg C/year.
Besides these five experiments, we carried out two additional runs based on the reference run RRR. In these two runs, designated RRR-CO and RRR-CH4, CO and methane emissions were fixed at their 1990 levels for all later years. The goal of these runs was to investigate the influence of CO emissions on predicted future concentrations of CO and related species, and on tropospheric chemistry in general.
Comparisons between observational data for CO and CH4 from stations at different latitudes and our modeled results showed that the model over-predicted somewhat surface CO and CH4 in the tropics and the Southern Hemisphere, but simulated it reasonably well in the Northern Hemisphere [Wang et al., 1998]. Overall, the model-simulated surface temporal trends of CO, CH4, and other trace gases fairly approximate zonal-average observations, building some confidence in the model's usefulness for predictions of trends.
Impact of Emissions on Future Carbon Monoxide
Our coupled model predicted a global-average mole fraction of tropospheric carbon monoxide in 2100 roughly double its present value. Different emission predictions cause clear differences among runs using identical assumptions for the remaining two parameters (i.e., the chemistry and climate model settings; see Figure 2a). For example, in the HRR run, the CO mole fraction reaches 198 ppb in the year 2100-respectively about 17 and 32 ppb higher than in the RRR and LRR runs in 2100.

Figure 2. Model predicted (a) tropospheric average mole fractions of CO in parts per billion (ppb) and (b) atmospheric lifetime of CO in month. Results from the HRR, RRR, and LRR runs are shown as functions of time.
As a result of increasing CO and CH4 emissions and the resultant depletion of hydroxyl radicals in the troposphere, the lifetime of carbon monoxide is predicted to lengthen significantly beyond its present value by the year 2100 (Figure 2b). The three runs using different emissions (HRR, RRR, and LRR) all predict a significant increase (0.6, 0.7, and 0.8 months respectively) above the present CO lifetime of 2 months. Higher emissions of CO and CH4 naturally induce a longer lifetime of CO (and obviously, of methane, as well) due to lower concentrations of hydroxyl radicals. This is despite the fact that higher NOx emissions help to offset the predicted reductions in OH radicals.
Detailed analyses suggest that the total chemical losses of CO also increase with time (Figure 3), despite the fact that predicted tropospheric concentrations of hydroxyl radicals in this model decrease 23-28% [Wang and Prinn, 1998a]. The differences of 80 Tg C/yr among chemical losses of CO in 2100 predicted from runs HRR, RRR, and LRR are much larger than the differences of 30 Tg C/yr among CO emissions used in these runs. This result is due to the fact that the production of CO from CH4 (through formaldehyde)-and therefore increasing methane emissions-dominates the predicted growth of atmospheric carbon monoxide. The annual growth rate (in percentage terms) of the CO mole fraction varied from lower than 1%/yr before the 1990s to around 1%/yr between 1990 and 2020, then to below 1%/yr again thereafter (Figure 4). This trend approximates well the trend derived from the predicted CH4 emissions used in our model. In contrast, the relative growth rates of predicted CO emissions are too small (<0.2%/yr) to explain the increase in CO mole fraction in the troposphere.
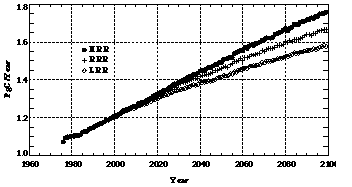
Figure 3. Model-predicted chemical losses of CO as functions of time. Data shown in the figure are from the HRR, RRR, and LRR runs and are in Pg C/year.
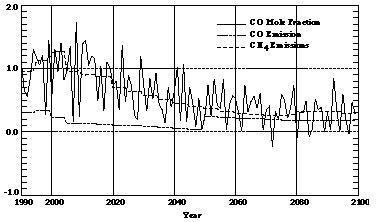
Figure 4. Growth rates of model predicted CO mole fractions, CO emissions, and CH4 emissions are shown as functions of time in percent per year. Note that the time step in the EPPA model is 5 years thus producing the small discontinuities in the emissions shown here.
Along with the change in lifetimes and abundance of CO and CH4 in the troposphere, a shift in the proportion of total OH consumed from CO toward methane occurs. Figure 5 shows that currently carbon monoxide consumes ~74% of hydroxyl radicals, while methane consumes the remaining 26%. However, our model predicts that in the year 2100, the proportion of hydroxyl radicals consumed by CO will fall to <69%, while that consumed by methane will rise to >30%. Note also that the chemical losses of both CO and CH4 through reactions with OH increase with time in our model runs. Consequently, emissions of methane are predicted to be more important factors for tropospheric chemistry in the year 2100 than they are currently.

Figure 5. Ratios of chemical losses of CO, L(CO), and of CH4, L(CH4), as functions of time. Results are from the HRR, RRR, and LRR runs.
Based on the predicted CO emissions used in this study, the future tropospheric abundance of CO will not differ significantly from its present level without a rapid increase in CH4 emissions. This conclusion is supported by results of the RRR-CO and RRR-CH4 runs, where CO or CH4 emissions are fixed at 1990 levels for all years following 1990. Figure 6a shows tropospheric-average mole fractions of CO predicted by the runs RRR-CH4, RRR, and RRR-CO. We find that restricting CO emissions to 1990 levels can reduce the mole fraction of CO in 2100 to about 20 ppb below the RRR prediction-a reduction much smaller, however, than the 74 ppb reduction predicted by Run RRR-CH4 which is based on constant post-1990 CH4 emissions. In the RRR-CH4 run, the predicted CO lifetime increases slightly after the 1990s, but eventually falls back to its present value after the year 2080 (Figure 6b). The CO lifetime predicted for Run RRR-CO (which has constant post-1990 CO emissions) closely approximates the reference run, RRR, in terms of both trends and absolute values.
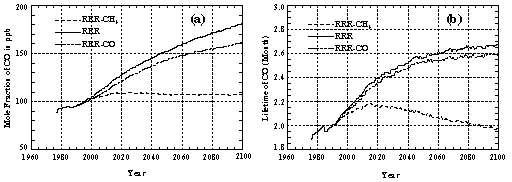
Figure 6. The same as in Figure 2, except using results from the RRR-CH4, RRR, and RRR-CO runs.
Tropospheric CO and Climate
Different assumptions about the aerosol radiative forcing intensity, the speed of oceanic uptake of CO2 and heat, and the sensitivity of the climate sub-model to doubled CO2, alter climate predictions and thus atmospheric chemistry, as well. For example, predictions of H2O (water vapor), a key species in atmospheric chemistry, differ among runs with different model assumptions (Figure 7). As a result, hydroxyl radical concentrations in these runs also differ, as do predicted mole fractions of CO (Figure 8a). A colder climate predicted in Run RLL leads to lower H2O and, hence, lower OH concentrations, and favors higher CO mole fractions compared to the other two runs. A colder climate also increases the CO lifetime more significantly (Figure 8b); specifically to 2.8 months in the year 2100, or about double the increase (from the present lifetime of 2 months) predicted in run RHH by the year 2100.
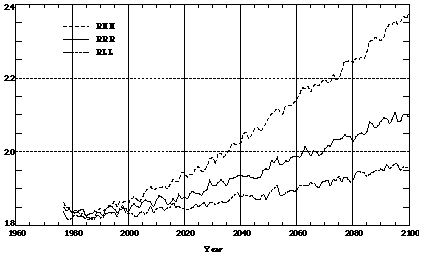
Figure 7. Model predicted tropospheric mixing ratios of H2O (water vapor) in g/kg. Shown are annual averaged values from the RHH, RRR, and RLL runs.
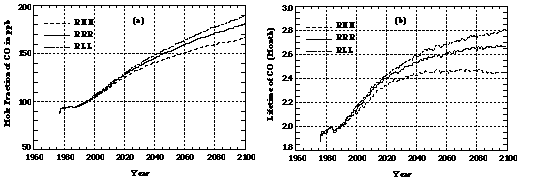
Figure 8. The same as in Figure 2, except using results from the RHH, RRR, and RLL runs.
We find that the annual chemical loss rates of CO due to reactions with hydroxyl radicals predicted in the RHH, RRR, and RLL runs are quite close to each other even though the OH concentrations derived from these runs differ (Figure 9). It suggests that contributions to the photochemical losses of CO attributable to changes in CO lifetime nearly offset those due to changes in CO abundance. This is because the short lifetime ensures that emissions and loss are always in near balance in all runs.
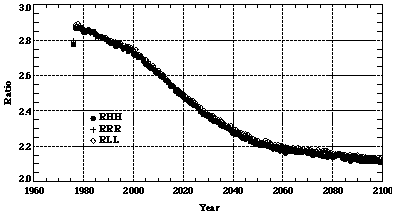
Figure 9. The same as in Figure 3, except using results from the RHH, RRR, and RLL runs.
Besides its role in tropospheric chemistry, carbon monoxide is the only internal atmospheric source of CO2. Figure 3 shows that CO2 production through the reaction of CO with OH is currently about 1.1 Pg C/yr, which is about 20% of estimated total current CO2 emissions [IPCC, 1996]. It is well known that CO and methane emissions are not negligible factors even in today's carbon dioxide budget. However, our work indicates that the fraction of CO contributions to the CO2 budget will become less significant as CO2 emissions reach much higher levels in the future. CO2 emissions are predicted to be about 20 Pg C/yr in 2100 [IPCC, 1996; Prinn et al., 1998], so that the contributions of carbon monoxide and methane emissions to CO2 would be less than 10% of the total CO2 input to the atmosphere by the end of the next century.
Another possible indirect effect of CO on the climate results from its influence on the tropospheric distribution of hydroxyl free radicals, and hence the oxidizing capacity of the troposphere. Growing CO and CH4 emissions can reduce OH levels in the troposphere [Wang and Prinn, 1998a] and hence the oxidation rate of methane and sulfur dioxide (the latter being a major sources of atmospheric aerosols).
Summary and Discussion
We have illustrated possible trends for atmospheric carbon monoxide in the next 100 years using our coupled atmospheric chemistry and climate model and three predictions from an economic development model for anthropogenic emissions. Complicated interactions among emissions, atmospheric chemistry, and climate impact the distribution and evolution of atmospheric CO. Based on predicted emissions of methane and carbon monoxide, our model predicts an increase in carbon monoxide over the next century, with the tropospheric mole fraction of CO in 2100 at double its present-day value. According to this study, methane emissions increase much more rapidly than CO emissions so that restricting CH4 emissions to current (1990) levels can prevent CO doubling from happening while controlling CO emissions alone cannot.
Our model runs show that high methane emissions will significantly deplete tropospheric OH, increase CO concentrations, and lengthen the lifetimes of many chemical species, including CO and CH4. For example, the CO lifetime in 2100 that predicted by our high-emission run (HRR) is about 2.8 months; currently, the CO lifetime is about 2 months or about 30% shorter. Emissions of CO are predicted to increase only about 0.16%/yr over the next century (Figure 1), so the impact of CO emissions on the CO abundance is less important than the impact of methane.
This study indicates that climate variations, especially those affecting H2O concentration, can influence atmospheric CO trends. Most interestingly, the atmospheric CO budget, which is affected by both CO and CH4 emissions, affects both the destruction of methane and production of CO2, ozone, and sulfate aerosols, and hence affects climate. The resultant climate changes (temperature, water vapor) can in turn modify the CO-CH4 budget. This offers a good example of how climate dynamics and atmospheric chemistry can interact.
Because our model omits non-methane hydrocarbons (NMHC), we may have underestimated CO and under- or overestimated hydroxyl radical concentrations (by excluding either ozone or H2O production or OH removal by NMHC, respectively). On the other hand, zonal-averaged models are typically biased to a NOx-poor atmosphere, so ozone production rates are possibly being underestimated due to this effect. More research is needed to investigate these uncertainties.
Acknowledgements
We thank our MIT colleagues Andrei Sokolov, Peter Stone, Xianming Xiao, Yuexin Liu, and Gary Holian for significant help and advice and for access to their models and model results. Jean Fitzmaurice provided help with emission calculations. Zili Yang, Henry Jacoby and their colleagues provided EPPA model outputs for anthropogenic emissions. Judy Stitt assisted in editing the manuscript.
This research is a part of the MIT Joint Program on the Science and Policy of Global Change, which is supported by a number of industrial sponsors [Asea Brown Boveri (Switzerland), Atlantic Richfield Company (USA), American Automobile Manufacture's Association (Chrysler, Ford, General Motors), British Petroleum Company (UK), Chevron Corporation (USA), Cyprus Amax Coal Company (USA), Electric Power Research Institute (USA), Exxon Corporation (USA), Mobil Corporation (USA), RWA/Rheinbraun (Germany), Saudi Arabian Oil Company, Shell Internationale Petroleum (The Netherlands), Statoil (Norway), Texaco Incorporated (USA), The Tokyo Electric Power Company (Japan)], by the U.S. Department of Energy (901214-HAR; DE-FG02-94ER61937; DE-FG02-93ER61713), U.S. National Science Foundation (9523616-ATM), U.S. National Oceanic and Atmospheric Administration (NA56GP0376), and U.S. Environmental Protection Agency (CR-820662-02), by the Royal Norwegian Ministries of Petroleum and Energy and Foreign Affairs, and by the G. Unger Vetlesen Foundation (USA).
References
Crutzen, P. J., and P. H. Zimmermann, The changing photochemistry of the troposphere, Tellus, 43AB, 136-151, 1991.
DeMore, W. B., S. P. Sander, D. M. Golden, R. F. Hampson, M. J. Kurylo, C. J. Howard, A. R. Ravishankara, C. E. Kolb, and M. JU. Molina, Chemical kinetics and photochemical data for use in stratospheric modeling. Evaluation Number 11, JPL Publication, Report No. 94-26, 1994.
Etheridge, D. M., G. I. Pearman, and P. J. Fraser, Changes in tropospheric methane between 1841 and 1978 from a high accumulation rate Antarctic ice core, Tellus, 44B, 282-294, 1992.
IPCC, Climate Change 1995: The Science of Global Change, Cambridge Univ. Press, Cambridge, UK, 572pp, 1996.
Khalil, M. A. K., and R. A. Rasmussen, Atmospheric methane: trends over the last 10,000 years, Atmos. Environ., 21, 2445-2452, 1987.
Khalil, M. A. K., and R. A. Rasmussen, Global cycle of CO-trends and mass balance, Chemosphere, 20, 227-242, 1990.
Khalil, M. A. K., and R. A. Rasmussen, The changing composition of the Earth's atmosphere, in Composition, Chemistry, and Climate of the Atmosphere, edited by Singh, H. B., pp. 51-87, Van Nostrand Reinhold, New York, 1995.
Klonecki, A., and H. Levy, II, Tropospheric chemical ozone tendencies in CO-CH4-NOy-H2O system: their sensitivity to variations in environmental parameters and their application to global chemistry transport model studies, J. Geophys. Res., 102, 21221-21237, 1997.
Levy, H., II, Normal atmosphere: Large radical and formaldehyde concentrations predicted, Science, 173, 141-143, 1971.
Novelli, P. C., K. A. Masarie, P. P. Tans, and P. M. Lang, Recent changes in atmospheric carbon monoxide, Science, 263, 1587-1590, 1994.
Prinn, R. G., H. Jacoby, A. Sokolov, C. Wang, X. Xiao, Z. Yang, R. Eckaus, P. Stone, D. Ellerman, J. Melillo, J. Fitzmaurice, D. Kicklighter, G. Holian, and Y. Liu, Integrated global system model for climate policy assessment: Feedbacks and sensitivity studies, Climatic Change, in press, 1998.
Rasmussen, R. A., and M. A. K. Khalil, Atmospheric methane in recent and ancient atmospheres: Concentrations, trends, and interhemispheric gradient, J. Geophys. Res., 89, 11599-11605, 1984.
Sze, N. D., Anthropogenic CO emissions: Implications for the atmospheric CO-OH-CH4 cycle, Science, 195, 673-675, 1977.
Thompson, A. M., and R. J. Cicerone, Atmospheric CH4, CO and OH from 1860 to 1985, Nature, 321, 148-150, 1986.
Wang, C., and R. G. Prinn, Combined effects of anthropogenic emissions and resultant climatic changes on atmospheric OH, submitted to Nature, 1998a.
Wang, C., and R. G. Prinn, Interactions among emissions, atmospheric chemistry, and climate change: Implications for future trends, submitted to J. Geophys. Res., 1998b.
Wang, C., R. G. Prinn, and A. Sokolov, A global interactive chemistry and climate model: Formulation and testing, J. Geophys. Res., 103, 3399-3417, 1998.
Yang, Z., R. S. Eckaus, A. D. Ellerman, and H. D. Jacoby, The MIT Emissions Prediction and Policy Analysis (EPPA) Model, MIT Joint Program on the Science and Policy of Global Change, Report No. 6, MIT, Cambridge, MA, 49pp, 1996.
| Top of page |
| |