Accurate Band Level Alignment Across Hybrid Interfaces
Understanding and controlling optoelectronic properties of hybrid interfaces that are composed of distinctly different materials  (i.e. self-assembled monolayers on a silicon surface) is of critical importance to the development of future optoelectronic technologies. The electronic energy-level alignment, the energetic offsets of near-gap states at the interface, is particularly crucial for designing optoelectronics with desirable attributes. We have developed an approach based on QMC calculations for obtaining the energy-level alignment at hybrid interfaces. For a representative organic-inorganic interface inclusion of accurate many-body effects from QMC calculations changes both quantitatively and qualitatively the DFT description of the energy-level alignment at the interface, as can be seen in the figure shown here.
(i.e. self-assembled monolayers on a silicon surface) is of critical importance to the development of future optoelectronic technologies. The electronic energy-level alignment, the energetic offsets of near-gap states at the interface, is particularly crucial for designing optoelectronics with desirable attributes. We have developed an approach based on QMC calculations for obtaining the energy-level alignment at hybrid interfaces. For a representative organic-inorganic interface inclusion of accurate many-body effects from QMC calculations changes both quantitatively and qualitatively the DFT description of the energy-level alignment at the interface, as can be seen in the figure shown here.
Coupled QMC/MD
We have developed an approach to provide accurate dynamical total energies by coupling stochastic, imaginary-time quantum Monte Carlo (QMC) with real-time ab initio molecular dynamics (AIMD) for ions. In this work, we showed that this coupling can be done efficiently for both ground and excited states with only a modest overhead to the AIMD method. 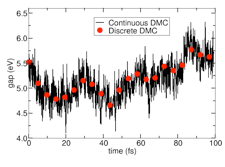 The initial method development was illustrated with application to small, high-temperature silicon clusters, the dissociation of a water molecule, and a short (2 ps) simulation of liquid water. The figure shown to the right illustrates the potential of the method. In it, the optical gap is computed on the fly for a Si5H12 cluster at 1000K (black curve) using the coupled approach. By contrast, the red points (error bars same size as symbols) correspond to discrete QMC calculations for each of the corresponding snapshots. The continuously sampled curve required 30X less computational time than the set of red data points. For more detail, please refer to: J. C. Grossman and L. Mitas, “Efficient Quantum Monte Carlo Energies for Molecular Dynamics Simulations,” Phys. Rev. Lett. 94, 056403 (2005).
The initial method development was illustrated with application to small, high-temperature silicon clusters, the dissociation of a water molecule, and a short (2 ps) simulation of liquid water. The figure shown to the right illustrates the potential of the method. In it, the optical gap is computed on the fly for a Si5H12 cluster at 1000K (black curve) using the coupled approach. By contrast, the red points (error bars same size as symbols) correspond to discrete QMC calculations for each of the corresponding snapshots. The continuously sampled curve required 30X less computational time than the set of red data points. For more detail, please refer to: J. C. Grossman and L. Mitas, “Efficient Quantum Monte Carlo Energies for Molecular Dynamics Simulations,” Phys. Rev. Lett. 94, 056403 (2005).
Geometry Optimization in QMC
For many interesting systems, such as transition metal systems, excited states, and weakly bound molecules, traditional first-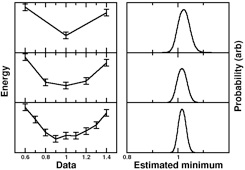 principles methods like density functional theory are not accurate enough for meaningful comparison to experiment. Quantum Monte Carlo has been used quite successfully to correct these deficiencies, but there is to date no reliable way to calculate forces on a general system. In addition, QMC energies come with statistical uncertainty, which makes direct energy minimization challenging. We are currently evaluating several methods to mitigate these issues and obtain highly accurate minimum energy structures.
principles methods like density functional theory are not accurate enough for meaningful comparison to experiment. Quantum Monte Carlo has been used quite successfully to correct these deficiencies, but there is to date no reliable way to calculate forces on a general system. In addition, QMC energies come with statistical uncertainty, which makes direct energy minimization challenging. We are currently evaluating several methods to mitigate these issues and obtain highly accurate minimum energy structures.



