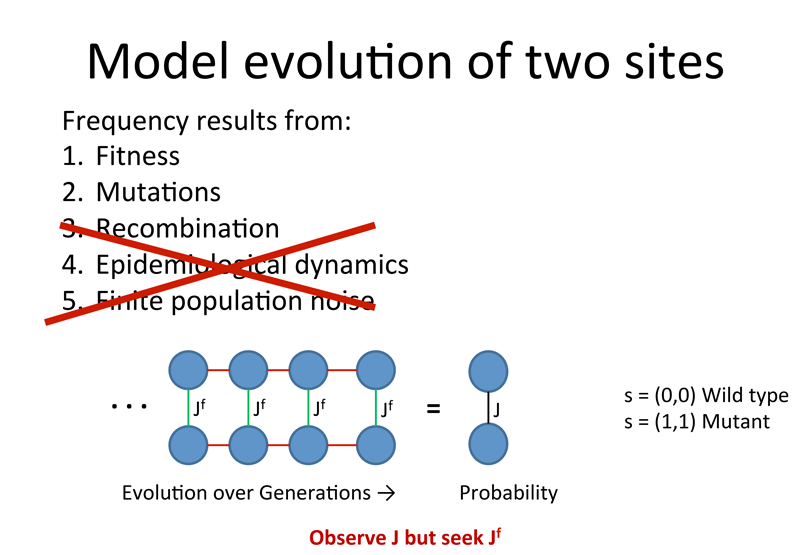
Protein Inhibotor resistance sites
![]() Hypothesis:
Hypothesis:
Single site mutations in response to drug carry large fitness cost.
![]() Test:
Test:

![]() Construct maximum entropy prevalence landscape (database www.hiv.lanl.gov)
Construct maximum entropy prevalence landscape (database www.hiv.lanl.gov)
- HIV-1 clade B Protease, removing insertions relative to HXB2
- Exclude all sequences after 1996, or with "protease inhibitor" in meta data [6701 from 757 patients]
- (Separate analysis with "drug naive" sequences)
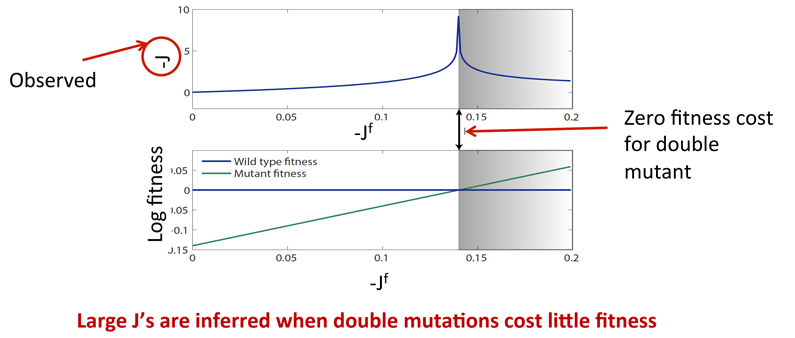
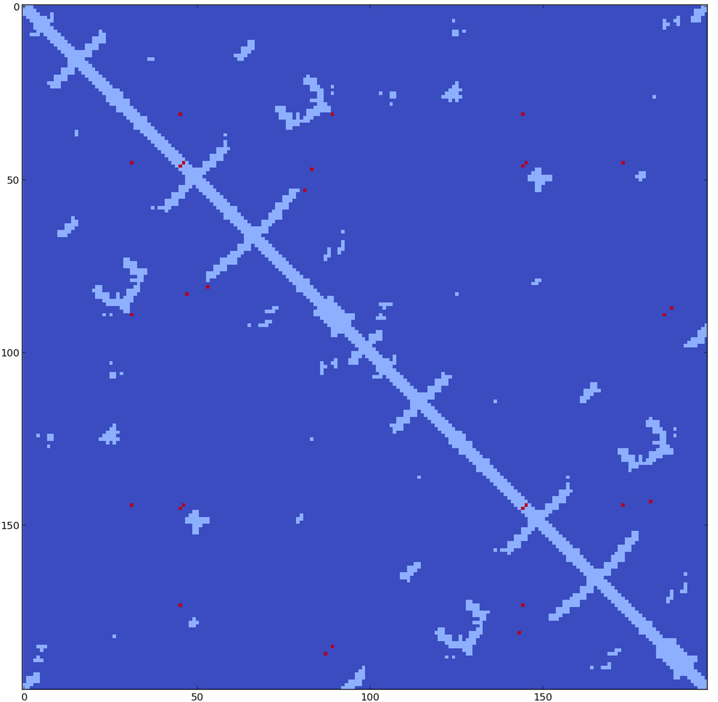
![]() Strong direct couplings indicate compensatory mutations of fitness close to wild type:
Strong direct couplings indicate compensatory mutations of fitness close to wild type:
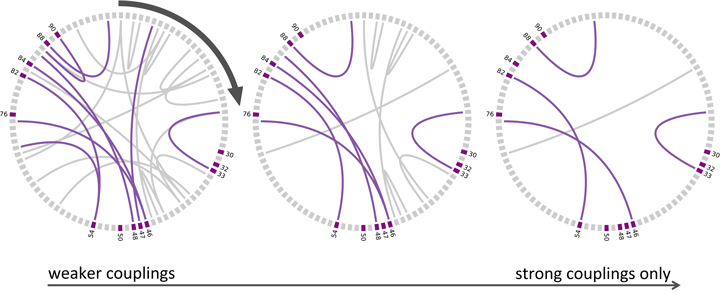
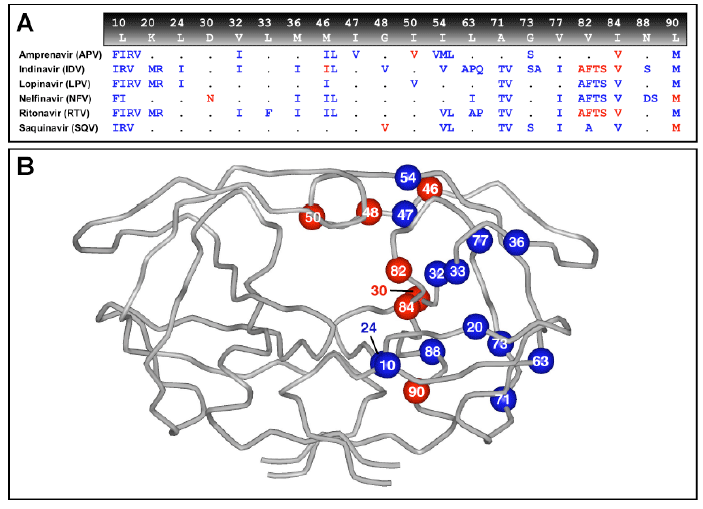
[PI Resistance sites, from M.E. Quiñones-Mateu1 and E.J. Arts, HIV Reviews 134 (2001)] [Rhee et al, NAR31, 298 (2003), Fig1]
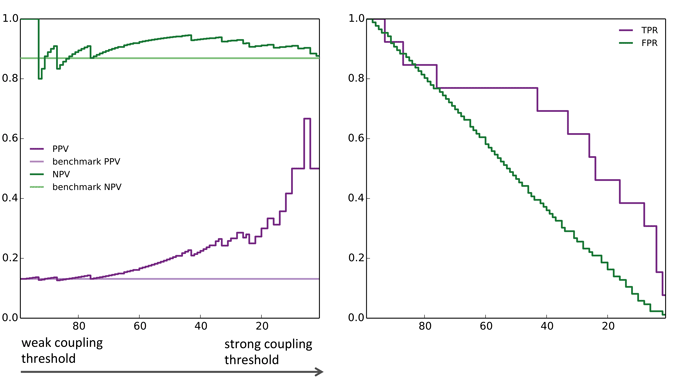
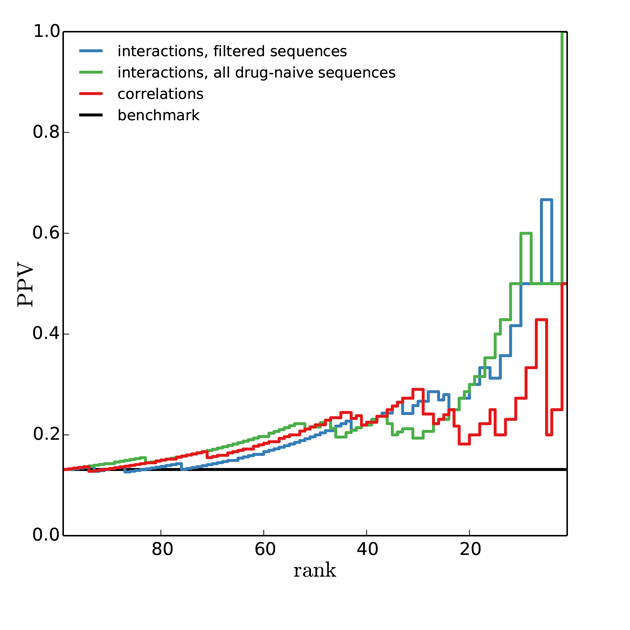
- PPV: Positive Prediction Value = Prob.[resistant is correct | predicted resistant ]
- NPV: Negative Prediction Value = Prob.[predicted non-resistant is correct | predicted non-resistant ]
- Larger PPV and NPV indicate more predictive power
- TPR: True Positive Rate = Prob.[predicted resistant is indeed resistant]
- FPR: False Positive Rate = Prob.[predicted resistant is non-resistant ]
- At large values of the threshold, drug resistance sites are identified with high probability
[T.C. Butler, J. Barton, M.K., A.K. Chakraborty (2014) & SI] (Fig. SI5)
![]() Many other approaches to predicting drug resistance sites:
Many other approaches to predicting drug resistance sites:
- Selection under treatment: L. Chen, A. Perlina, CJ Lee, J Viriology 78, 3722 (2004)
- Supervised learning: CW Cao et al, Drug Discovery Today 10, 521 (2005)
- Structural modelling: N Beerenwinkel et al, PNAS 99, 8271 (2002)
- ...
{kind=link}
{kind=link}
{kind=link}