CHAPTER 3: REGULATORY MOTIF DISCOVERY
3.1. Introduction
Regulatory motifs are short nucleotide sequences typically upstream of genes that are used to control the expression of genes, dictating under which conditions a gene will be turned on or off. Direct identification of regulatory elements is more challenging than that of genes. Such elements are typically short (6-15 bp), tolerate some degree of sequence variation and follow few known rules. To date, the majority have been found by experimentation, such as systematic mutation of individual promoter regions; the process is laborious and unsuited for genome-scale analysis.
Computational analysis of single genomes has been successfully used to identify regulatory elements associated with known sets of related genes7-9. These methods typically search for frequently-occurring sequence patterns at various distances upstream of coordinately expressed genes, and will be further described in chapter 4. They are however limited by the experimental information available, and hence do not permit a comprehensive direct identification of regulatory elements43.
Comparative genomics offers various approaches for finding regulatory elements. The simplest approach is to perform cross-species sequence alignment to find phylogenetic footprints, regions of unusually high conservation. This approach has long been used to study promoters of specific genes in many organisms10,12,44-46 and recently was applied across the entire human and mouse genomes19. The genome alignments of the four Saccharomyces species can similarly be used to study each yeast gene, to help define promoters and other islands of intergenic conservation (Figure 3.2).
Our interest was to go beyond inspection of individual islands of conservation to construct a comprehensive dictionary of regulatory elements used throughout the genome. We investigated the conservation properties of known regulatory motifs and used the insights gained to design an approach for de novo discovery of regulatory motifs directly from the genome.
In this chapter, we develop and apply methods for genome-wide motif discovery. We compare our results to a database of experimentally validated regulatory motifs and rediscover virtually all previously known motifs. In chapter 4 we develop methods for inferring a candidate function for the motifs discovered making use of biological knowledge about genes, and in chapter 5 we explore their combinatorial interactions.
3.2. Regulatory motifs
The current knowledge of gene regulation is based on focused experimental studies of specific examples. The deletion of a transcription factor was shown to disrupt the use of its target genes. Regulatory elements were identified in genetic screens through function-disrupting mutations that reside outside of a protein-coding ORF. Systematic mutagenesis of a particular promoter region (also known as promoter bashing) and testing the resulting effect on gene expression has been used to identify functional blocks in upstream regions of genes. To identify regulatory motifs at a nucleotide level, footprinting methods can be used. These methods expose the bound region to DNA damaging agents that degrade unbound nucleotides, leaving a ‘footprint’ of the transcription factor on the bound and thus protected nucleotides. Finally, even higher resolution information is obtained through crystal structures of transcription factors bound to DNA. These different methods have produced lists of bound sites for each of a small number of well-studied transcription factors.
|
IUB |
Nucleotides |
Name |
[pA,pc,pG,pT] |
|
A |
A |
Adenine |
[1, 0, 0, 0] |
|
C |
C |
Cytosine |
[0, 1, 0, 0] |
|
G |
G |
Glutamine |
[0, 0, 1, 0] |
|
T |
T |
Tyrosine |
[0, 0, 0, 1] |
|
S |
C or G |
Strong |
[0, ˝, ˝, 0] |
|
W |
A or T |
Weak |
[˝, 0, 0, ˝] |
|
R |
A or G |
PuRine |
[˝, 0, ˝, 0] |
|
Y |
C or T |
pYrimidine |
[0, ˝, 0, ˝] |
|
M |
A or C |
aMino group |
[˝, ˝, 0, 0] |
|
K |
G or T |
Keto group |
[0, 0, ˝, ˝] |
|
B |
C or G or T |
Not A |
[0, ⅓, ⅓, ⅓] |
|
D |
A or G or T |
Not C |
[⅓, 0, ⅓, ⅓] |
|
H |
A or C or T |
Not G |
[⅓, ⅓, 0, ⅓] |
|
V |
A or C or G |
Not T |
[⅓, ⅓, ⅓, 0] |
|
N |
A, C, G or T |
aNy base |
[Ľ, Ľ, Ľ, Ľ] |
|
Table 3.1. Degenerate nucleotide code. |
The sites bound by these factors exhibit sequence similarities that reveal the binding specificity of each factor, and can be represented in a regulatory motif. Representations for these motifs range from consensus sequences listing the nucleotides involved in binding, to weight matrices and graphical models. Consensus sequences or sequence profiles are the simplest such representation, giving a list of possible bases for each position in the bound site. Some positions are strict and require the presence of a particular nucleotide, others allow for degeneracies. These can be represented compactly using the IUB standard one-letter code (Table 3.1). More complex representations can be used allowing for more detail in the binding specificity. A weight matrix representation of a motif of length L assigns weight vector wi = [wA, wC, wG, wT] to every position i between 1 and L. The binding strength of a sequence can be scored against a weight matrix by simply adding up the corresponding scores for each position. In a probabilistic framework, the weights can represent the relative frequencies of each nucleotide in real motifs, multiplying across the corresponding weights gives the probability that a sequence s matches the motif represented by m. Alternatively, if log probabilities are used instead, summing across the matrix gives the corresponding log probability. This probability can be compared to the probability of obtaining s by chance, to obtain a log-likelihood ratio that the sequence matches the motif. Both consensus sequences and weight matrices model the binding contributions of nucleotide position as independent. More complex Bayesian representations for motifs can be used to capture pairwise and multiple dependencies between positions. As the models become more complex however, the increased power comes at a cost, increasing the number of parameters and possibly overfitting data.
Transcription factors have evolved different ways to contact the DNA double helix, and these are reflected in different types of regulatory motifs. Some factors make one long contact with the DNA helix recognizing between 6 and 8 positions, some of which can be degenerate. One such example is the Mbp1 transcription factor involved in the timing of events such as DNA replication during cell division and recognizes the motif ACGCGT. Other factors contact the DNA at two different points, resulting in motifs with two cores, separated by a stretch of unspecified bases. For example, the binding site recognized by Abf1, a general transcription factor involved in silencing and replication, recognizes the motif RTCRYNNNNNACGR. The DNA-binding domains of other factors are made of two identical parts (and hence called homodimers), contacting each other and each contacting the DNA helix. The two parts recognize identical sequences, but on opposite strands, and hence result in motifs that are reverse palindromes of themselves. One such example is the Gal4 factor involved in galactose metabolism, recognizing CGGNNNNNNNNNNNCCG, namely CGG on one strand spaced by 11 nucleotides (one full turn of the double helix) from its reverse complement, CCG.
3.3. Extracting signal from noise
Computationally, discovering regulatory motifs amounts to extracting signal from noise. When the motifs searched are expected to be more frequent than other patterns of the same length, one can apply discovery algorithms such as Expectation Maximization (EM) or Gibbs sampling (and others reviewed in ref 9). These were pioneered by Lawrence and coworkers47, and made popular in software programs like MEME7,48, AlignACE8,49,50 or BioProspector51. More recent work has extended these methods to incorporate phylogenetic footprinting45,52-54. These methods separate the motif discovery problem in two sub-problems. (1) Given a set of starting coordinates i1, …, in in each of the sequences, construct the optimal matrix representation for a motif that starts at each of these positions. (2) Given a matrix representation for a motif m, find the starting positions of the best matches for that motif in each of the sequences. These algorithms start with a random assignment for the start positions and infers the best matrix, then iterates to improve the assignment of start positions to better match the motif. EM algorithms choose the optimal assignment for each of these rounds of iteration. Gibbs sampling algorithms instead sample amidst the best start positions. Both algorithms converge as long as the motif searched is actually frequent in the sequences searched, since probabilistically, the algorithms will be likely to sample these motifs in their iterative steps, and upon sampling them will converge to include them.
These methods have typically been applied to the upstream sequences of small sets of genes, but are not applicable to a genome-wide discovery. Instead, k-mer counting methods have been used to find short sequences that occur more frequently in intergenic regions, as compared to coding regions in a genome-wide fashion43. However, these typically find very degenerate sequences (such as poly-A or poly-T) and have shown limited power to separate regulatory motifs from the mostly non-functional intergenic regions. This is largely due to the small number of functional instances of regulatory motifs, as compared to the large number of non-functional nucleotides. The discovery of regulatory motifs still relies heavily on extensive experimentation.
Comparative genomics provides a powerful way to distinguish regulatory motifs from non-functional patterns based on their conservation. In this chapter we first study conservation properties of known regulatory motifs. We use these to construct three tests to detect the genome-wide signature of motif-like conservation. We use these tests to detect all significant patterns with strong genome-wide conservation, constructing a list of 72 genome-wide motifs. We compare this list against previously identified regulatory motifs and show that our method has high sensitivity and specificity, detecting most previously known regulatory motifs, but also a similar number of novel motifs. In chapter 4, we assign candidate functions to these novel motifs, and in chapter 5, we study their combinatorial interactions.
3.4. Conservation properties of known regulatory motifs
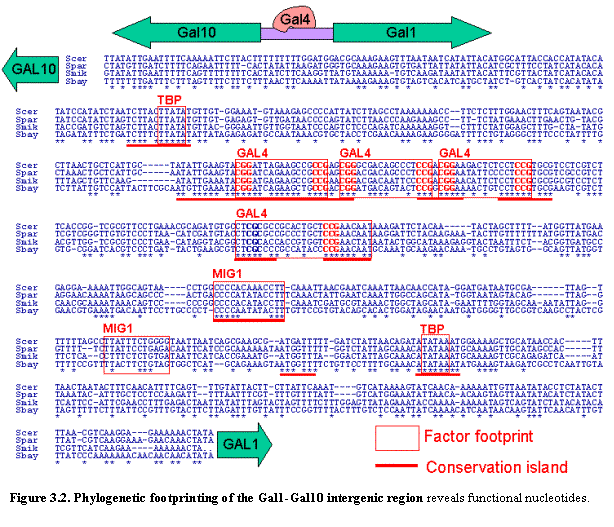
We first studied the binding site for one of the best studied transcription
factors, Gal4, whose sequence motif is CGG(N)11CCG (which contains 11
unspecified bases). Gal4 regulates genes involved in galactose utilization,
including the GAL1 and GAL10 genes that are divergently
transcribed from a common intergenic region (Figure 3.2). The Gal4 motif occurs
three times in this intergenic region, and all three instances show perfect
conservation across the four species. In addition, there is a fourth,
experimentally validated binding site55 for Gal4 that differs from
the consensus by one nucleotide in S.
cerevisiae. This variant site is also perfectly preserved across the
species.
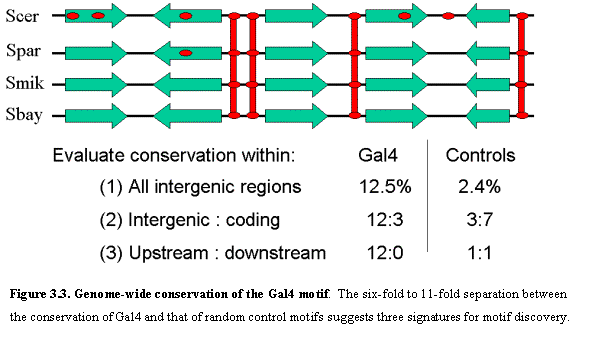
We then examined the frequency and conservation of Gal4 binding sites across
the aligned genomes (Figure 3.3). In S. cerevisiae, the Gal4 motif occurs 96
times in intergenic regions and 415 times in genic (protein coding) regions.
The motif displays certain striking conservation properties. First, occurrences of the Gal4 motif in
intergenic regions have a conservation rate (proportion conserved across all
four species) that is ~5-fold higher than for equivalent random motifs (12.5%
vs. 2.4%). Second, intergenic occurrences of the Gal4 motif are more frequently
conserved than genic occurrences (12.5% vs. 3%). By contrast, random motifs are
less frequently conserved in intergenic regions than genic regions (3.1% vs.
7.0%), reflecting the lower overall level of conservation in intergenic
regions. Thus, the relative conservation rate in intergenic vs. genic regions
is ~11-fold higher for Gal4 than for than random motifs. Third, the Gal4 motif
shows a higher conservation rate in divergent vs. convergent intergenic regions
(those that lie upstream vs. downstream of both flanking genes); no such
preferences is seen for control motifs. These three observations suggest
various ways to discover motifs based on their conservation properties (see
conservation criteria below).
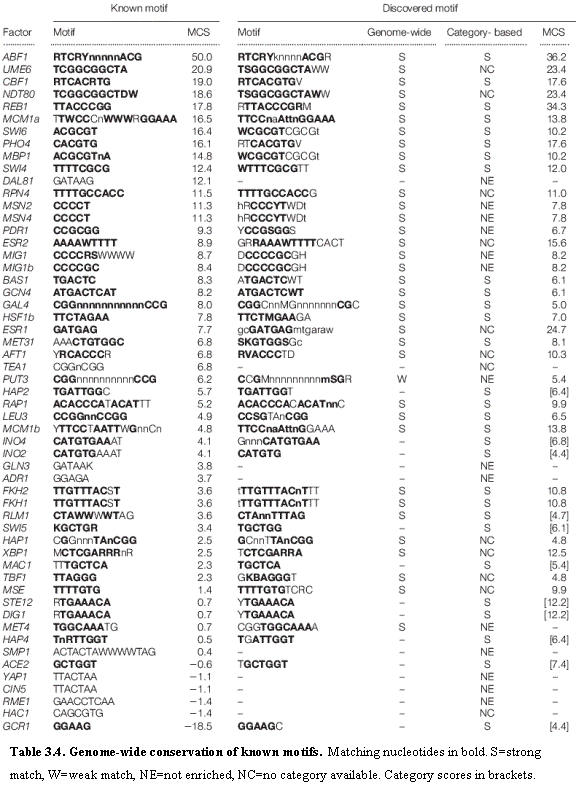
We extended these observations by assembling a catalog of 55 known regulatory
sequence motifs (Table 3.4), by starting with two public databases (SCPD56,57 and YTFD58) and curating the entries to
select those with the best support in the literature. We defined a Motif Conservation Score (MCS) based on the
conservation rate of the motif in intergenic regions. To evaluate the Motif Conservation Score (MCS) of a motif m of given length and degeneracy, we
compared its conservation ratio to that
of random patterns of the same length and degeneracy. We first computed the table F
containing the relative frequencies of two-fold and three-fold degenerate bases,
given the S. cerevisiae nucleotide
frequencies (.32 for A and T, .18 for C and G). For example, W=[AT] (.32*.32) is a more likely two-fold
degenerate base than Y=[CT] (.18*.32).
We then selected 20 random intergenic loci in S. cerevisiae. For each of
these loci, we used the order of nucleotides at that locus together with the
order of degeneracy levels in m to construct a random motif. If the first character of m
was two-fold degenerate and the first nucleotide at the selected locus was A,
we picked a two-fold degenerate base containing A (W, R or M), their relative
frequencies dictated by F, and continued for every character
of m. We then counted conserved and non-conserved
instances of each of the 20 generated control patterns and computed r,
the log-average of their conservation rates.
We then counted the number of conserved and non-conserved intergenic
instances of m, and computed the binomial probability p of observing the two
counts, given r. We finally reported
the MCS of the motif as a z-score corresponding to p, the number of standard
deviations away from the mean of a normal distribution that corresponds to tail
area p. Nearly all of these sequence motifs are
binding sites of known transcription factors.
Most of the known motifs show extremely strong conservation, with 60%
having MCS ≥ 4 (which is substantially higher than expected by chance).
Some of the motifs, however, show relatively modest MCS. These motifs may be
incorrect, suboptimal or not well conserved.
3.5. Genome-wide motif discovery
Our methodology for genome-wide motif discovery involves first identifying conserved mini-motifs and then using these to construct full motifs (Figure 3.5). Mini-motifs are sequences of the form XYZn(0-21)UVW, consisting of two triplets of specified bases interrupted by a fixed number (from 0 to 21) of unspecified bases. Examples are TAGGAT, ATAnnGGC, or the Gal4 motif itself. The total number of distinct mini-motifs is 45,760, if reverse complements are grouped together.
Conserved mini-motifs are evaluated according to three conservation criteria (CC1-3), based on our observations about the properties of the Gal4 motif. In each case, conservation rates are normalized to appropriate random controls. CC1 (Intergenic conservation) evaluates the conservation rate of a mini-motif in intergenic regions. CC2 (Intergenic-genic conservation) evaluates the stronger conservation in intergenic regions as compared to genic regions. CC3 (Upstream-downstream conservation) evaluates the different conservation of a mini-motif when it occurs upstream vs. downstream of a gene.
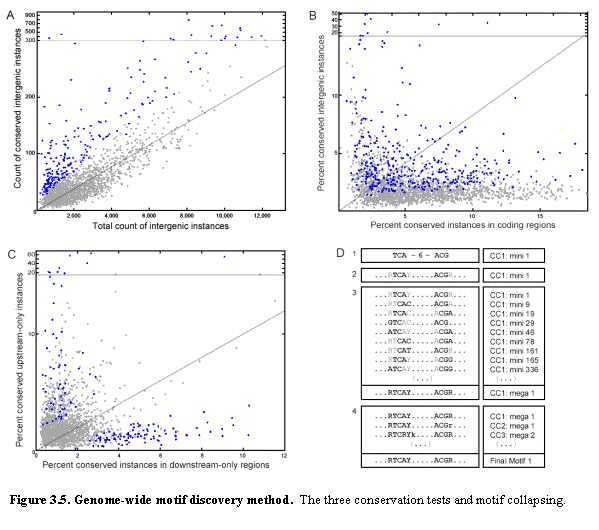
CC1: Intergenic conservation. We searched for mini-motifs that show a
significant conservation in intergenic regions. For every mini-motif, we counted ic the number of
perfectly conserved intergenic instances in all four species, and i
the total number of intergenic instances in S.cerevisiae. We found that the two counts seem linearly
related for the large majority of patterns (Figure 3.5 panel A), which can be
attributed to a basal level of conservation r given the total
evolutionary distance that separates the four species compared. We estimated the ratio r as the log-average of
non-outlier instances of ic/i within a control set of all
motifs at a given gap size. We then
calculated for every motif the binomial probability p of observing ic
successes out of i trials, given parameter r.
We assigned a z-score S to every motif corresponding to
probability p. This score is
positive if the motif is conserved more frequently than random, and negative if
the motif is diverged more frequently than random. We found that the distribution of scores is symmetric around zero
for the vast majority of motifs. The
right tail of the distribution however is much further than the left tail,
containing 1190 motifs more than 5 sigma away from the mean, as compared to 25
motifs for the left tail. By comparing
the two counts, we estimated that 94% of these 1190 motifs are non-random in
their conservation enrichment.
CC2: Intergenic-genic conservation. We searched for motifs that are preferentially conserved in intergenic regions, as compared to coding regions. In addition to ic and i (see previous section), we counted the number of conserved coding instances gc, and the number of total coding instances g, for every mini-motif. We observed the ratio of conserved instances that are intergenic a=ic/(ic+gc), and compared it to the total ratio of motif instances that are intergenic b=i/(i+g). Not surprisingly, we found that typically b=25% of all motif instances appeared in intergenic regions, which account for roughly 25% of the yeast genome. Similarly, only a=10% of conserved motif instances appeared in intergenic regions, which reflects the lower conservation of intergenic regions. To correct for this typical depletion in intergenic conservation, we estimated a correction factor f=a/b for mini-motifs of similar GC-content. Then for a given mini-motif, the proportion of all instances found in intergenic regions and the correction for the lower conservation of intergenic regions together gave us r=f*i/(i+g), the expected ratio of conserved intergenic instances for that motif. We evaluated the binomial probability p of of observing at least ic conserved instances in intergenic regions and ic+gc conserved instances overall, given the expected ratio r. As in CC1, we computed a z-score S for every motif and found a distribution centered around zero for the large majority of motifs, and a heavier right tail. We selected 1110 motifs above 5 sigma and estimated that 97% are non-random as compared to only 39 motifs below -5 sigma.
CC3: Upstream-downstream conservation. We searched for motifs that are differentially conserved in upstream regions and downstream regions. We defined upstream-only intergenic regions in divergent promoters that are upstream of both flanking ORFs, and downstream-only intergenic regions in convergent 3’ terminators that are downstream of both flanking ORFs. We then counted uc and u, the conserved and total counts in upstream-only regions, and similarly dc and d in downstream-only regions. We found that upstream-only and downstream-only regions have similar conservation rates, and the ratios uc/u and dc/d are both similar to ic/i for the large majority of motifs. We thus used a simple chi-square contingency test on the four counts (uc,u,dc,d) to find motifs that are differentially conserved. We found 1089 mini-motifs with a chi-square value of 10.83 or greater, which corresponds to a p-value of .001. Given the multiple testing of 45760 mini-motifs, we estimated that roughly 46 will show such a score by chance and that 96% of the selected motifs will be non-random.
The conserved mini-motifs are then used to construct full motifs (Figure 3.5). They are first extended, by searching for nearby sequence positions showing significant correlation with a mini-motif. The extended motifs are then clustered, merging those with substantially overlapping sequences and those that tend to occur in the same intergenic regions. Finally, a full motif is created by deriving a consensus sequence (which may be degenerate). Motifs are typically degenerate, and a single full-motif can be responsible for multiple strong mini-motifs. We now describe methods to recover the full motifs and their degeneracy.
We extended each mini-motif selected by searching for
surrounding bases that are preferentially conserved when the motif is
conserved. We used an iterative
approach adding at every iteration one base that maximally discriminates the
neighborhood of conserved motif instances from the neighborhood of
non-conserved motif instances. The
added base was selected from fourteen degenerate symbols of the IUB code (A, C,
G, T, S, W, R, Y, M, K, B, D, H, V).
When no such symbol separated the conserved and non-conserved instances
with significance above 3 sigma, we terminated the extension. Figure 3.5 panel D shows the top-scoring
mini-motif found in CC1 (Row 1), and the corresponding extension (Row 2). We found that many mini-motifs have the same
or similar extensions, and we grouped these based on sequence similarity. We measured the similarity between two
motifs as the number of bits in common in the best ungapped 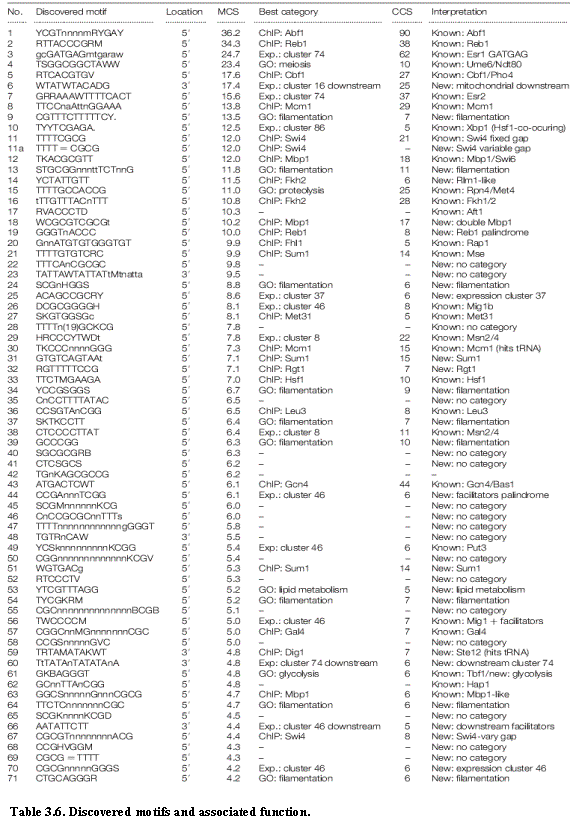
alignment of the two motifs, divided by the minimum number of bits contained in
either motif. Based on the pairwise
motif similarity matrix, we clustered the extended motifs hierarchically,
collapsing two groups if the average similarity between their member motifs was
at least 70%. We then computed a
consensus sequence for every cluster of extended motifs, resulting into a
smaller number of mega-motifs for each test (332 for CC1, 269 for CC2 and 285
for CC3). Row 3 shows the first 9
members of the top cluster in CC1, and the resulting mega-motif. Finally, we merged mega-motifs based on
their co-occurrence in the same intergenic regions (Row 4). We computed a hypergeometric co-occurrence
score between the intergenic regions hit by each mega-motif and again collapsed
these hierarchically. We computed a
consensus for every cluster, and iterated the co-occurrence-based collapsing
step (results not shown). We obtained
fewer than 200 distinct genome-wide motifs.
Each full motif is assessed for genome-wide conservation by calculating
its MCS, and those motifs with MCS ≥ 4 are retained. Each full motif was also tested for
enrichment in upstream vs. downstream regions, by comparing its conservation
rate in divergent vs. convergent intergenic regions.
3.7. Results and comparison to known motifs
The vast majority of the 45,760 possible mini-motifs show no distinctive conservation pattern. However, ~2400 mini-motifs show high scores by one or more of these criteria (Figure 3.5 panels A, B, C). There is substantial overlap among the mini-motifs produced by the three criteria, with about 50% of those found by one criterion also found by another.
The conserved mini-motifs give rise to a list of 72 full motifs having MCS ≥ 4 (Table 3.6). We omit full motifs with low MCS scores, and those that overlap tRNA genes and may be due to secondary RNA structure. Most of the motifs show preferential enrichment upstream of genes, but six are enriched downstream of genes. These 72 discovered motifs, found with no prior biological knowledge, show strong overlap with 28 of the 33 known motifs having MCS ≥ 4. They include 27 strong matches and 1 weaker match. The 72 discovered motifs also contain matches to 8 of the 22 known motifs with MCS < 4. In these cases, the comparative analysis identified closely related motifs that have higher conservation scores than the known motifs and occur largely at the same genes; these may represent a better description of the true regulatory element. Comparative genomic analysis thus automatically discovered 36 motifs with matches to most of the known motifs (65% of the full set, 85% of those with high conservation). It also identified 42 additional ‘novel’ motifs not found in our list of known motifs. In the next chapter, we develop methods to understand these novel motifs and assign a candidate function to each of them.
3.8. Conclusion
Motif discovery amounts to extracting small sequence signals hidden within largely non-functional intergenic sequences. This problem is difficult in a single genome where the signal-to-noise ratio is very small. Previous methods have thus been limited to discovering motifs within small sets of genomic regions. We have conducted a genome-wide exhaustive search for all regulatory motifs. We produced a list of 72 strongly conserved motifs, that includes most previously identified motifs. This ability to directly discover regulatory motifs drastically changes our view of gene regulation. Instead of a case-by-case study, we can now observe complete views of all regulatory building blocks. Our method has re-discovered most previously known regulatory motifs without use of any prior biological function. It should theoretically be applicable to any genome for which no experimental data is available. Additionally, in yeast, we can use the biological information to discover the function of the discovered motifs. We can also use biological function to discover additional motifs. These two goals will be the topic of the next chapter.