CHAPTER 4:Ā REGULATORY MOTIF FUNCTION
4.1. Introduction
In response to environmental changes, a single transcription factor can induce the expression of all genes necessary to fulfill a particular function, such as galactose import and utilization.Ā These genes are typically scattered throughout the genome and targeted by the presence in their upstream regions of a specific regulatory motif recognized by the factor.Ā This regulatory motif will be enriched in the upstream regions of these genes, namely it will occur more frequently in these regions than expected by chance as compared to the rest of the genome.
This enrichment of regulatory motifs in functionally related sets of genes can be used in two ways.Ā Given a gene set, an associated motif can be found by searching the upstream intergenic regions for short patterns occurring at an unusual frequency.Ā Alternatively, given a novel motif whose function is unknown, an associated gene set can be found by testing a number of previously defined gene sets (categories) for enrichment.Ā
In a single genome, motifs occur frequently by chance, and hence the enrichment observed is sometimes not sufficient to perform either of these two tasks with high sensitivity and specificity.Ā With multiple aligned genomes at hand, most spurious motif instances can be eliminated and the enrichment should become more pronounced.Ā We can use this increased power to assign a candidate function to the motifs discovered in the previous chapter and to discover additional motifs in a category-specific way.
In this chapter, we present methods to distinguish biologically meaningful motif instances under selective pressure from non-functional motif instances.Ā We assign candidate functions to the genome-wide motifs discovered in the previous chapter and find that the majority of discovered motifs show a significant functional enrichment.Ā We also present a new method to discover additional regulatory motifs associated with functional categories.Ā For known factors, we find that our category-based discovery method has great sensitivity and specificity, finding concise binding sites even when previous methods fail.Ā For all 354 categories tested, we find that only a small number of motifs are found and these are shared, reused across categories.
4.2. Constructing functionally-related gene sets.
In yeast, a number of genome-wide experiments have resulted in functional groupings of genes into gene sets.Ā These represent possibly co-regulated groups, constructed from gene expression, transcription factor binding and protein function.
Microarray technology enables the simultaneous measurement of gene expression levels for all 6000 annotated yeast genes on a single array.Ā Such arrays contain thousands of spots (one for every gene), each containing multiple single-stranded nucleotide probes complementary to the corresponding predicted yeast gene.Ā When cell extract is washed on the array, the single-stranded mRNA transcripts present in the cell hybridize (bind) by complementarity to the appropriate spots in the array.Ā The level of hybridization can be measured by first fluorescently labeling the mRNA transcripts and then measuring the level of fluorescence on each spot using a laser scanner.Ā The higher the hybridization measured at a spot, the higher is the inferred level of mRNA expression for that gene. These genome-wide experiments have been repeated for hundreds of experimental conditions and expression profiles have been constructed for every gene, describing its expression levels in each condition.Ā These profiles can then be clustered computationally59, typically by their pairwise correlation coefficients, to obtain sets of transcriptionally coordinated sets of genes.
Another technology, ChIP, has recently been applied to the genome-wide level to observe the binding locations of a transcription factor across the genome60,61.Ā This technology enables the specific targeting of a transcription factor of interest, in order to pull it out of a cell extract.Ā Pulling a transcription factor also selects for the DNA fragments that it is bound to.Ā A researcher can then hybridize these fragments against an array containing probes for promoter regions, and infer which regions are bound by the transcription factor.Ā Current technologies target transcription factors by either constructing an antibody specific to the factor, or by appending to the transcription factor a tag to which an antibody already exists (antibodies are molecules used by our immune system to recognize specific proteins of invading agents like viruses or bacteria; hence the name of Chromatin Immuno-Precipitation abbreviated as ChIP, referring to the use of antibodies to cause the chromatin bound by a factor to precipitate with the factor when this one is pulled).Ā The DNA is fragmented before precipitation and only a few hundred bases surrounding the bound site are typically pulled.
Genes can also be grouped into functional categories, based on the experimentally determined function of the proteins they encode.Ā The function of thousands of yeast genes has been experimentally determined (to various degrees of precision).Ā The scientific papers that describe these functions have been manually curated by the Saccharomyces Genome Database (SGD) group, generating a vast repository of knowledge.Ā This knowledge has been classified hierarchically into Gene Ontology (GO) information or MIPS62, using a unified language that crosscuts species and organism boundaries.Ā This hierarchy groups at each internal node genes of related function, from the most specific to the most general, in categories such as æmeiotic DNA double-strand break processingÆ, æcell cycleÆ, or æmetabolismÆ.Ā Genes of related function will sometimes be part of the same metabolic pathway, required simultaneously for the correct sequence of chemical modifications of a metabolite, and hence likely to be co-regulated.Ā Similarly, proteins that are part of the same protein complex are likely to be co-regulated, since they are required simultaneously for the correct assembly of the protein complex.Ā Experimental methods similar to ChIP can be used to detect protein complexes63:Ā an antibody specific to one of the proteins in the complex is used to pull the entire complex out of cell extract;Ā the complex pulled is then fractionated at specific residues and the charge/weight combination of the fragments obtained by Mass Spectroscopy are used to find the precise set of amino acids in the fragment and the corresponding proteins that can result in such amino acid subsets.
4.3. Assigning a function to the genome-wide motifs
We used the biological knowledge captured in these sets of functionally related genes to assign function to the 72 genome-wide motifs discovered in the previous chapter.Ā Since motifs can be degenerate and sometimes conserved in only a subset of the species, we first developed methods to score conserved motif instances.Ā We then evaluated the overlap between the set of intergenic regions with motif scores above a given cutoff, and each functionally-related set of genes.Ā We found a strong overlap with functional sets for most of the genome-wide motifs, and discover novel motif functions.
We used a probabilistic representation to detect conserved motif instances.Ā We interpret every genome-wide motif m of length L as a probabilistic model, generator of sequences of length L over the alphabet {A,C,G,T}.Ā We then evaluated for every genome position, the probability that the sequence was generated by motif m, and compared this to the probability that the sequence was generated at random, given the ratio of A,C,G,T in the genome.Ā We evaluated each species in turn, to obtain a total number of bits in the alignment.Ā Since gaps may exist in the alignment, we did not evaluate the motif match directly on the alignment.Ā Instead, we evaluated the motif in the ungapped sequence of each species in turn, and translated the motif start coordinates based on the alignment.Ā To avoid evaluating each of 12 million start positions in the yeast genome against the motif, we first hashed the four genomes for rapid lookup, and subsequently only search those intergenic regions that contain k-mers in the motif searched.Ā To allow for degenerate matches, we also search for k-mers with one or two degeneracies from the query motif. We then used a simple threshold t and obtain the list of all intergenic regions containing conserved instances of the motif with score at least t.Ā These instances are either upstream of downstream of each flanking gene, depending on its transcriptional orientation.Ā We could thus generate an æupstreamÆ list of genes that contain these conserved instances in their upstream regions, and a corresponding ædownstreamÆ list of genes. We compared the overlap between each upstream and downstream gene list against each set of functionally related genes.Ā
We did not expect a perfect overlap where every gene in a category would contain the motif and every gene outside the category would not contain the motif.Ā On one hand, we expected discrepancies due to experimental errors, incomplete annotations and artifacts of the clustering algorithms.Ā But even with perfect data, discrepancies arise from molecular processes that cross-cut functional categories, transcription factor binding that is dependent on additional protein-protein interactions or chromatin structure, expression clusters that are controlledĀ by multiple transcription factors.Ā At the same time, much like spurious motif instances can occur in a single species when motifs are short and degenerate, even conserved motif instances can occur by chance, although less frequent.Ā Similarly, functional motif instances may appear diverged due to alignment errors, or may have genuinely diverged across the species compared.
Thus, we evaluated
the overlap between motif presence and functional information
probabilistically.Ā Assume that m
genes contain the motif and r genes belong to a particular functional
category.Ā At random, if the motif is
independent from the category, we expect the same proportion of genes to
contain motif instances both inside and outside the category.Ā The probability of observing a deviation
from that ratio can be evaluated using the hypergeometric distribution, described
in the appendix.Ā If k genes are
observed in the overlap between the two sets, and n genes are present in
the yeast genome, we calculate a P-value that the enrichment is observed at
random as the hypergeometric sum for all values of kÆ that are greater
or equal to k.Ā Since we were evaluating the
overlap of each motif against a large number of candidate functional
categories, we use a Bonferroni correction for multiple hypothesis testing.
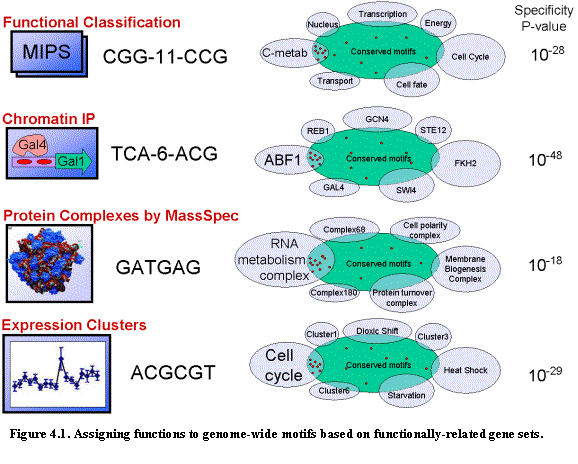
We applied these ideas to the motifs we discovered in our genome-wide
search.Ā As a control, we used the Gal4
motif (Figure 4.1). Given the biological role of Gal4, we considered the set of
genes annotated to be involved in carbohydrate metabolism (126 genes according
to the Gene Ontology (GO)64 classification) with the set
of genes that have a Gal4 binding motif upstream. The intergenic regions
adjacent to carbohydrate metabolism genes comprise only 2% of all intergenic
regions, but 7% of the occurrences of the Gal4 motif in S. cerevisiae (3.5-fold enrichment) and 29% of the conserved
occurrences across the four species (15-fold enrichment).Ā These results suggest that a function of the
Gal4 motif could be inferred from the function of the genes adjacent to its
conserved occurrences.Ā Such putative
functional assignments can be useful in directing experimentation for
understanding the precise function of a motif.Ā
Novel functions for genome-wide motifs
We compared each of the 72 motifs against a collection of 318 yeast gene categories based on functional and experimental data described earlier.Ā These categories consist of 120 sets of genes defined with a common GO classification in SGD64; 106 sets of genes whose upstream region was identified as binding a given transcription factor in genome-wide chromatin immunoprecipitation (ChIP) experiments61; and 92 sets of genes showing coordinate regulation in RNA expression studies59.Ā To measure how strongly the conserved occurrences correlated with the regions upstream (or downstream) of a particular gene category.Ā We require a hypergeometric score of at least 10-5 to judge an overlap as significant, after accounting for testing of multiple categories.Ā Most of the 36 discovered motifs that correspond to known motifs showed strong category correlation. Categories with the strongest correlation included those identified by ChIP with the transcription factor known to bind the motif, although many other relevant categories were identified. Of the 42 novel motifs, 25 show strong correlation with at least one category and thus can be assigned a suggestive biological function (Table 3.6).
Some motifs appear to define previously unknown binding sites associated with known transcription factors. Motif 32 is likely to be the binding site for Rgt1, which regulates genes involved in glucose transport65; the motif occurs upstream of many such genes, including appearing five times upstream of HXT1, which encodes a high-affinity glucose transporter. Motifs 21, 31 and 51 are all associated with genes whose upstream regions are bound by Sum1, a transcriptional repressor of genes involved in meiosis. The first motif has been previously reported (MSE)66, but the latter two are novel and occur near genes whose products are involved in chromatin silencing and transcriptional repression.
Other motifs do not match regions bound by known transcription factors, but show strong correlation with functional categories. Motif 9 occurs upstream of genes involved in nitrogen metabolism, including amino acid and urea metabolism, nitrogen transport, glutamine metabolism and carbamoyl phosphate synthesis.Ā Motif 25 is enriched among co-expressed genes (expression cluster 37) whose products function in vesicular traffic and secretion, including GDP/GTP exchange factors essential for the secretory machinery, clathrin assembly factors and many vesicle and plasma membrane proteins. Motifs 9, 13, 26, 34, 37 may play a role in filamentation.Ā They are all enriched in genes co-regulated during environmental changes, involved in signaling and budding and bound by transcription factors involved in filamentation, such as Phd1.
Six motifs show higher conservation downstream of ORFs.Ā Some of these may be in the 3Æ untranslated region of a transript and play a regulatory role in mRNA localization or stability.Ā The strongest (Motif 6 and 67) is found at genes whose product localizes to the cytosolic translational machinery, the mtDNA translational machinery or the mitochondrial outer membrane. Downstream motifs are also found enriched in a group of genes repressed during environmental stress (Motif 60 with expression cluster 37) and a group of genes involved in energy production (Motif 66 with expression cluster 46).
Two motifs (Motif 11a and Motif 69) show variable gap spacing, suggesting a new type of degeneracy within the recognition site for a transcription factor complex.Ā Motif 11a corresponds closely to the known motif for Swi4 (Motif 11) but is interrupted by a central gap of 5, 7 or 9 bases; these variant motifs all show strong correlation with genes bound by Swi4 in ChIP experiments.
4.4. Discovering additional motifs based on gene sets
We next explored whether additional motifs could be found by searching specifically for conservation within individual gene categories.Ā We selected mini-motifs based on their enrichment in specific categories and extended them to full motifs.Ā We first evaluated our motif discovery method for ChIP experiments of factors with known motifs, and we found high sensitivity and specificity.Ā We then searched for novel motifs in all 318 functional categories and discovered novel motifs.
The enrichment of regulatory motifs found in co-regulated gene sets has been
the primary motivation for motif discovery algorithms such as MEME, AlignAce or
BioProspector.Ā These algorithms
typically search for frequently occurring motifs within the set and
subsequently evaluate the significance of the enrichment observed based on the
overall frequency of the motifs throughout the genome.Ā Thus, they search for motifs that are
frequent within the set, and filter out those that are also frequent outside
the set.Ā We select for both criteria
simultaneously by choosing mini-motifs based directly on their category
enrichment score.Ā We counted the
conserved instances within the category (IN), and the conserved instances
outside the category (OUT).Ā We estimated
the ratio p=IN/(IN+OUT) that we should expect for the category, based on
the entire population of mini-motifs.Ā
We then calculated the significance of an observed enrichment as the
binomial probability of observing IN successes out of IN+OUT trials given the
probability of success p.Ā We
assigned a z-score to each mini-motif, as described in the genome-wide
search.Ā We extended those mini-motifs
of z-score at least 5 sigma by searching for neighboring conserved bases that
increase the specificity.Ā We finally
collapsed motifs of similar extension based on sequence similarity.
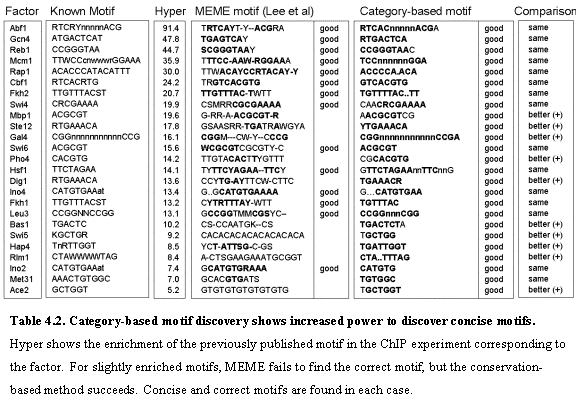
We first evaluated our ability to detect the 43 known motifs for which ChIP experiments61 had been performed with the transcription factor that binds the motifs. For each category defined by the ChIP experiment, we undertook category-based motif discovery.Ā Strong category-based motifs were found in 29 cases and these invariably corresponded closely to the known motifs (Table 4.2). These include 11 cases in which the motif had not been found by genome-wide motif discovery, suggesting that a category-based approach can be more sensitive in some cases.Ā No strong category-based motifs were found for the remaining 14 known cases, including 7 cases in which genome-wide analysis yielded the known motif.Ā Analysis of these 14 known motifs showed that none were, in fact, enriched in the ChIP-based category.Ā This may reflect errors in the known motifs in some cases and imperfect ChIP data in others. Genome-wide analysis may simply be more powerful than category-based analysis in some instances.Ā In all, 46 of the 55 known motifs were found by either genome-wide or category-based analysis. The remaining 9 cases may reflect true failures of the comparative genomic analysis or errors in the known motifs.Ā
We compared our results to the motifs discovered by MEME in a single species as reported in Lee et al61.Ā Our method showed stronger sensitivity in discovering all motifs for which the ChIP experiment indeed contained the correct motif.Ā Additionally, the method showed strong specificity in the motifs discovered:Ā the motifs were short and concise, and closely matched the published consensus.Ā On the contrary, MEME failed to find the true motif in a number of cases, and when a motif was found it was generally obscured by a number of surrounding spurious bases that are not reported in the known motifs.Ā Thus, we successfully used the additional information that comes from the multiple alignment to improve category-based motif discovery with very satisfactory results.Ā By comparing multiple species, the signal becomes stronger.Ā It allows the search to focus on the conserved bases, eliminating most of the noise.Ā Table 1 summarizes the results. For each factor, we show the published motif, the hypergeometric enrichment score of the motif within the category (Hyper), the motif discovered by MEME and a quality assessment, the motif discovered by our method, as well as the corresponding category-based score and a quality assessment, and finally the comparison of our method to MEME.Ā The performance of MEME degrades for less enriched motifs, but we consistently find the correct motif.

We then applied the approach to all 318 gene categories.Ā A total of 181 well-conserved motifs were
identified, with many of these being equivalent motifs arising from multiple
categories. Merging such motifs resulted in 52 distinct motifs, of which 43
were already found by the analyses described above. The remaining 9 motifs
represent new category-based motifs (Table 4.3), including the following.
Three novel motifs are associated with genes that are bound by the transcription factors Rap1, Ste12 and Cin5, respectively. Rap1 is known to bind incomplete or degenerate instances of the published motif and the new motif may confer additional specificity. The motif associated with Ste12 is the known binding site for the partner transcription factor Tec1, suggesting that Ste12 binding is strongly associated with its partner under the conditions examined. Similarly, the novel motif associated with Cin5 may be that of a partner transcription factor.Ā Three novel motifs are associated with the GO category for carbohydrate transport, fatty-acid oxidation and glycolysis-glycogenesis, respectively. Three novel motifs are associated with an expression cluster (cluster 37) that includes many genes involved in energy metabolism and stress response.
4.7. Conclusion
Category-based motif discovery contributes only a modest number of additional motifs beyond those found by genome-wide analysis.Ā This confirms the relatively small number of regulatory motifs in yeast.Ā A limited count is surprising given the large number of coordinately transcribed processes in yeast.Ā The versatility of fine-grain yeast regulation may be rooted in a combinatorial control of gene expression, which will be the topic of the next chapter.