CHAPTER 5: COMBINATORIAL REGULATION
5.1. Introduction
We also used the comparisons to understand combinatorial interactions between regulatory motifs.Ā A simple view of gene regulation where each environmental response is regulated by a dedicated transcription factor would require as many transcription factors and regulatory motifs as there are molecules and environmental changes.Ā This is however not the case.Ā It is estimated that only 160 transcription factors exist in the yeast genome, but yeast cells contain thousand of co-regulated sets of genes.Ā This discrepancy requires a different model of gene regulation that goes beyond a one-to-one correspondence between regulatory motifs and cellular processes.
Our results from the previous chapter indeed point to a model where specific motif combinations are responsible for different cell responses.Ā We saw that a single motif is typically involved in the control of many processes, and that a single process is typically enriched in multiple regulatory motifs.Ā Furthermore, we saw that different processes were enriched in different combinations of regulatory motifs.Ā Protein-protein interactions between the multiple factors bound upstream of every gene may dictate the specific combination of conditions under which the gene will be expressed. Understanding the combinations of regulatory motifs that are biologically meaningful, and the changing target gene sets may explain the versatility of eukaryotic gene regulation using only a small number of regulatory building blocks.
In this chapter, we develop methods to reveal the combinatorial control of gene expression.Ā We construct a global motif interaction map, simply based on proximity of conserved motif pairs without requiring biological knowledge of gene function.Ā We then present evidence for the changing functional specificities of the motif combinations discovered.Ā Finally, we show the genome-wide effect of motif combinations on gene expression change.
5.2. Motifs are shared, reused across functional categories
We saw in the previous chapter that the motifs discovered across different categories largely overlapped.Ā Each motif was discovered on average in three different categories.Ā This overlap is certainly to be expected between functionally related categories such as the chromatin IP experiment for Gcn4, the expression cluster of genes involved in amino acid biosynthesis, as well as the GO annotations for amino acid biosynthesis, all of which are enriched in the Gcn4 motif, the master regulator of amino acid metabolism.
More surprisingly however, different transcription factors are often enriched in the same motif (which may be due to cooperative binding), and the same motif appears enriched in multiple expression clusters and functional categories.Ā For example, Cbf1, Met4, and Met31 share a motif, and so do Hsf1, Msn2 and Msn4; Fkh1 and Fkh2; Fhl1 and Rap1; Ste12 and Dig1; Swi5 and Ace2;Ā Swi6, Swi4, Ash1 and Mbp1.Ā Also, a single motif involved in environmental stress response is found repeatedly in numerous expression clusters, and in functional categories ranging from secretion, cell organization and biogenesis, transcription, ribosome biogenesis and rRNA processing.Ā
Hence, the set of regulatory motifs that are specific to one functional category seems limited.Ā This can hamper category-based motif discovery methods:Ā no category will be enriched in a single motif, and no motif will be enriched in a single category.Ā Additionally, there are a number of experimental limitations to a category-based approach.Ā For example, the expression clusters we have used, although constructed over an impressive array of experiments, are still limited to the relatively few experimental conditions generated in the lab.Ā Additionally, the functional categories we used are limited to the few well-characterized processes in yeast, and the molecular function of more than 3000 ORFs remains unknown.Ā
A genome-wide approach presents a new and powerful paradigm to understanding the dictionary of regulatory motifs.Ā By discovering in an unbiased way the complete set of conserved sequence elements, we now have the building blocks to subsequent analyses of regulation.Ā To understand the full versatility of gene regulation, we now turn to understanding the combinatorial code of motif interactions.Ā We first show that motif combinations can change the specificity of target genes, not in an additive, but in a combinatorial way.Ā We then present methods to discover interacting motifs from the genome-wide co-occurrence of their conserved instances, without making use of functional information.Ā We then show that the interactions found are meaningful.
5.3. Changing specificity of motif combinations.
The effect of motif sharing a reuse can be additive or combinatorial.Ā An additive effect simply adds the effect of the co-occurring transcription factors.Ā For example, if each of two factors induces the expression of a gene, and both bind to a particular region, then their effect would be a doubly increased level of transcription for that gene.Ā A combinatorial effect can be more complex.Ā Namely, the combination of two factors may repress expression for a gene, even though either of the factors alone induces its expression.
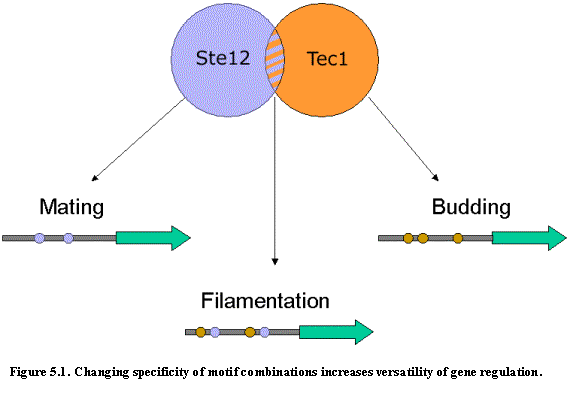
Similarly, we should find that transcription factor combinations show different
functional specificities than either of the transcription factors alone (Figure
5.1).Ā We study here the gene category
enrichment of two transcription factors that are known to bind to DNA
cooperatively:Ā Ste12 and Tec1.Ā We considered three types of regions:Ā those containing Tec1 motifs but no Ste12
motifs, those containing Ste12 motifs but no Tec1 motifs, and those containing
both Ste12 and Tec1 motifs.Ā We then
intersected these three types of regions against the gene sets described
previously.Ā
We found that the regions that contain only the conserved Ste12 motif are enriched for genes involved in mating and pheromone response, while those that contain conserved occurrences of both the Ste12 and Tec1 motifs are enriched for genes involved in filamentous growth. These computational observations are consistent with recent elegant work showing genome-wide evidence that Ste12 and Tec1 indeed cooperate during starvation to induce filamentation-specific genes68. We also found that regions that contain only conserved occurrences of the Tec1 motif are enriched for genes involved in budding and cell polarity, suggesting that Tec1 has functions that do not require cooperative binding with Ste12.
5.4. Genome-wide motif co-occurrence map.
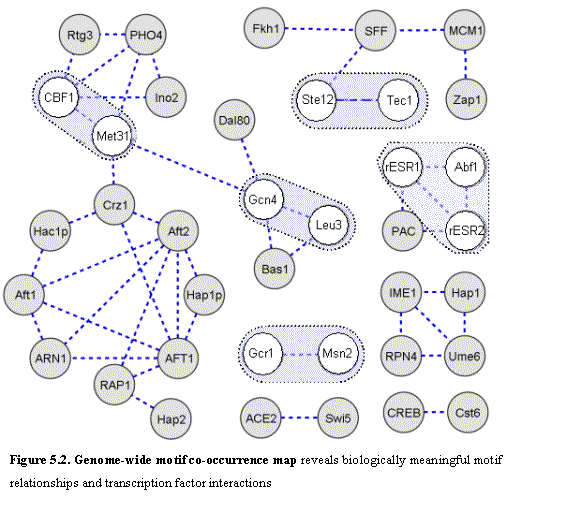
We next address the question of discovering these motif interactions in a
genome-wide fashion.Ā Protein-protein
interactions between cooperatively binding transcription factors require that
they bind in proximity upstream of their target genes.Ā The regulatory motifs recognized by these
factors should therefore co-occur in these intergenic regions of cooperative
binding.Ā The spatial orientation and
physical distance between these motifs may vary across different genes, the
varying distances being compensated by DNA bending that can bring the two sites
in proximity.Ā However, motif
interactions do not typically cross gene boundaries, that are enforced by
chromatin packaging and larger physical distances from one intergenic region to
the next.Ā Thus, co-occurrence of
regulatory motifs in the same intergenic regions might be a good indicator of
interacting transcription factors.
Using the comparison of the four species, we observed the genome-wide co-occurrence patterns of regulatory motifs (Figure 5.2).Ā We searched for motifs that occur in the same intergenic regions more frequently than one would expect by chance.Ā We computed the probability of seeing at least k regions in common when one motif is found in m regions and the other motif is found in r regions, given a total of n intergenic regions using the hypergeometric distribution.Ā
Without using any functional information of gene categories, we found a number of significant motif interactions.Ā These group motifs together into complex motif co-occurrence networks that may form the basis for studying combinatorial regulation of gene expression.Ā These are not apparent in a single genome, where functional instances of the motif are overwhelmed by a much larger number of random occurrences.Ā Cross species conservation greatly decrease this random noise and reveals biologically meaningful correlations.
5.5. Results.
We outlined here a number of biologically significant connections in the motif co-occurrence map.Ā The combinatorial effect between Ste12 and Tec1 was indeed observed at the genome-wide level.Ā The Ste12 and Tec1 motifs show clear correlation, with about 20% of regions having a conserved occurrence of one also having a conserved occurrence of the other. This enrichment is not apparent when considering S. cerevisiae alone.
The motif co-occurrence map reveals a number of biologically meaningful interactions.Ā (a) About 60% of regions containing conserved motifs for the transcription factor Leu3 (which regulates branched-chain amino-acid biosynthesis) also contain conserved motifs for Gcn4 (a general factor regulating amino acid biosynthesis, as well as many other processes).Ā (b) About 46% of regions containing conserved motifs for the transcription factor Met31 also contain conserved occurrences of Cbf1. In fact, Cbf1 (which is involves in DNA bending) is known to physically interact and cooperate with the MET regulatory complex. (c) About 34% of regions containing a conserved Gal4 motif also contain a conserved Mig1 motif. In this case, the correlation reflects antagonistic interaction. Gal4 induces galactose metabolism genes in presence of galactose, but Mig1 represses galactose metabolism in presence of glucose.Ā (d) Pairwise co-occurrence connects a group of five motifs: Msn2/4 (general stress response), Rlm1 (response to cell-wall stresses), Pdr1 (pleiotropic drug resistance), Tea1 (Ty element activator) and Tbf1 (Telomere-binding factor).Ā This suggests a possible link between various stress responses and adaptive changes at the genome level69.Ā
Many additional correlations are seen among known and novel motifs and can be pursued experimentally and computationally to construct comprehensive co-occurrence networks.Ā These can provide information valuable in deciphering biological pathways in yeast.
5.6. Conclusion.Ā
In this chapter, we provide methods to discover meaningful combinatorial interactions between regulatory motifs in a genome-wide way.Ā Motif combinations can change the functional specificity of downstream motifs, and regulate a large number of processes using only a small number of regulatory motifs.Ā This combinatorial nature of yeast regulation allows for a robust and modular regulatory network to adapt to changing environmental conditions.Ā It is possible that additional regulatory motifs are added to the network, modulated by the more stable master regulatory motifs.Ā We can further pursue these ideas to understand the rewiring of regulatory networks across evolutionary time.Ā This may be one of many subtle ways of rapid evolutionary change outlined in the next chapter.