CHAPTER 6:Ā EVOLUTIONARY CHANGE
6.1. Introduction
In previous chapters, we used the stronger conservation of functional elements across related species for the direct identification of genes and regulatory motifs.Ā However, the species compared are not identical.Ā They live in different environments and are subject to different pressures for survival.Ā In the short evolutionary time that separates them, they have undergoneĀ a number of evolutionary changes to adapt to their respective environments.Ā
In comparative genomics, both similarities and differences of the species compared can reveal important biological principles.Ā Focusing on the similarities gives us a view of a core cell whose functionality has remained unchanged since the common ancestor of the species.Ā Focusing on the differences gives us a dynamic view of a changing genome, and the mechanisms evolved for rapid adaptation to changing environments.Ā
In this chapter, we focus on the mechanisms of evolutionary change that have become apparent in our comparisons.Ā We show that the ambiguities in gene correspondence found in chapter 1 are localized in rapidly evolving telomeric regions at the chromosome endpoints.Ā We also show that non-telomeric changes in gene order are due to either the inversion of a chromosomal segment (containing fewer than 20 genes) or reciprocal exchanges of chromosomal arms.Ā For both types of events, the sequences at the breakpoints suggest specific mechanisms of chromosomal change.Ā We observed few differences in gene content between the species, suggesting that phenotypic differences may be due to more subtle effects like protein domain changes and changes in gene regulation.Ā Finally, we observed rapidly and slowly evolving genes:Ā at one end of the spectrum, we found evidence of positive selection for rapid change in membrane adhesion proteins, suggesting a small number of mechanisms of rapid change;Ā we also found genes that were surprisingly strongly conserved suggesting new hypotheses for their function.
6.2. Protein family expansions localize at the telomeres.
In the previous chapters, we used unambiguous ORFs and intergenic regions to discover conserved coding and regulatory elements in the yeast genome.Ā In this chapter, we use ORFs with ambiguous correspondence to determine regions of rapid change.Ā
We marked the chromosomal location of all S. cerevisiae ORFs that are ambiguous in at least one species.Ā We then constructed ambiguity clusters when two or more ambiguous ORFs within 16kb of each other.Ā We counted the number of ambiguities in each cluster, counting more than one ambiguities for an ORF whose correspondence was ambiguous in more than one species.Ā Only 32 clusters were found containing more than two ambiguities.Ā We ignored two clusters due to regions of low coverage in S. mikatae and one cluster corresponding to a previously described inversion.
Most
of the ambiguities are strikingly clustered in telomeric regions (Figure 6.1).
More than 80% fall into one of 32 clusters of two or more genes (average size
~18 kb, together comprising ~4% of the genome), which correspond nearly
perfectly to the 32 telomeric regions of the 16 chromosomes of S. cerevisiae.
Only one telomeric region lacks a cluster and only one cluster does not lie in
telomeric regions in S. cerevisiae: it is a recent insertion of a segment that
is telomeric in the other three species. The rapid structural evolution in the
telomeric regions can also be observed at the gene level. The gene families
contained within these regions (including the HXT, FLO, PAU, COS, THI, YRF
families) show significant changes in number, order, and orientation. The
regions also harbor many novel sequences, including protein-coding
sequences.Ā Finally, the telomeric
regions have undergone 11 reciprocal translocations across the species.Ā
Together, these features define relatively clear boundaries for the telomeric regions on all 32 chromosome arms, with sizes ranging from ~7 kb to ~52 kb on chromosome I-R. The extraordinary genomic churning occurring in these regions - and the telomeric localization of environment adaptation protein families - together probably play a key role in rapidly creating phenotypic diversity over evolutionary time.Ā A high degree of variation in telomeric gene families has also been reported in P. falsiparum69, the parasite responsible for malaria, and is related to antigenic variation.

6.3. Chromosomal rearrangements mediated by specific sequences.Ā

ĀĀOutside
of the telomeric regions, few genomic rearrangements are found relative to S.
cerevisiae (Figure 6.2).Ā To
discover these, we considered consecutive unambiguous matches, marking all
changes in gene spacing, gene orientation, and off-synteny matches between
scaffolds and orthologous S. cerevisiae chromosomes.Ā We found that changes in gene spacing are
typically associated with transposon insertions and associated novel genes, as
well as tandem duplications.Ā Virtually
all changes in gene orientation typically affect between 2 and 10 consecutive
ORFs and can be traced to one of 16 multi-gene inversions.Ā The majority of off-synteny matches involve
a single ORF and only 20 involve more than 2 consecutive ORFs.Ā Virtually all single-gene off-synteny
matches were contained within ancient duplication blocks of Saccharomyces
as described in 70 and http://acer.gen.tcd.ie/~khwolfe/yeast/nova/.Ā These probably represent previously
duplicated genes that were differentially lost in different species, rather
than a DNA break in one of the two lineages, as was previously noted in 71.Ā Off-synteny matches that involve more than two genes from the
same chromosome correspond to one of 20 chromosomal exchanges.
S.
paradoxus shows no reciprocal translocations, 4 inversions and 3 segmental
duplications. S. mikatae shows 4 reciprocal translocations and 13 inversions.
S. bayanus has 5 reciprocal translocations and 3 inversions. The results
confirmed four recently reported reciprocal translocations in these species,
identified by pulsed-field gel electrophoresis72, and identified four additional reciprocal translocations that had been
missed.Ā The sequence at the chromosomal
breakpoints suggested the possible mechanism that underlie the rearrangements.
Strikingly, the 20 inversions are all flanked by tRNA genes in opposite
transcriptional orientation and usually of the same isoacceptor type; the
origins of inversions in recombination between tRNA genes has not previously
been noted. The reciprocal translocations occurred between Ty elements in seven
cases and between highly similar pairs of ribosomal protein genes in two cases;
the implication of Ty elements in reciprocal translocation is consistent with
previous reports44,71-73. One segmental duplication involves ædonorÆ and ærecipientÆ regions
that are descendants of an ancient duplication in the yeast genome70. Differential gene loss of anciently duplicatedĀ genes has been previously reported74, but this is the first observation of a recent re-duplication event
within anciently duplicated regions.
6.4. Small number of novel genes separate the species
We found a very small number of genes unique to one species and absent in the others. We noted above that S. cerevisiae contains 18 genes for which we could not identify orthologs in any of the other species, of which 7 encode ≥ 200 aa. These may be species-specific genes in S. cerevisiae, but alternatively could simply reflect gaps in the available draft genome sequences. ĀĀĀĀĀ
This uncertainty does not arise, however, in the reverse direction in identifying genes in the related species that lack an ortholog in S. cerevisiae. We found a total of 35 such ORFs encoding ≥ 200 aa (with the minimum length chosen to ensure that these are likely to represent valid genes). The list includes 5 genes unique to S. paradoxus, 8 genes unique to S. mikatae (two of which are 99% identical) and 19 genes unique to S. bayanus (three of which form a gene family with ≥ 90% pairwise identity). There is also one gene represented by orthologous ORFs found in the latter two species only and one represented by orthologous ORFS in all three related species.
These species-specific ORFs are notable with respect to both function and location. The majority (63%) can also be assigned biological function on the basis of strong protein-sequence similarity with genes in other organisms. Most involve sugar metabolism and gene regulation (including one encoding a silencer protein). The majority (69%) are found in telomeric regions and an additional set (17%) are immediately adjacent to Ty elements; these locations are consistent with rapid genome evolution.
A curious coincidence was noted in the region between YFL014W and YFL016W in S. cerevisiae. In the orthologous regions in all four species, we find a species-specific ORF in every case (165, 111, 136 and 228 aa), but these four ORFs show little similarity at the protein level. The amino acid sequence has been disrupted by frame-shifting indels, but a long ORF has been maintained in each case. The explanation for this phenomenon is unclear, but may prove interesting.
6.5. Slow evolution suggests novel gene function.
With sequence alignments at millions of positions across the four species, it is possible to obtain a precise estimate of the rate of evolutionary change in the tree connecting the species.
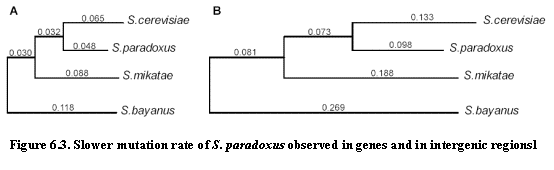
One notable observation is the difference in substitution rate between S.
cerevisiae and S. paradoxus (Figure 6.3).Ā Using S. bayanus as an
outgroup, the substitution rate is about 67% lower in the lineage leading to S. paradoxus.Ā This observation is consistent regardless of the measure of
evolutionary change:Ā mutations,
insertions, deletions measured across intergenic regions, genes or degenerate
nucleotides in coding sequence all point to the same discrepancy.Ā Hence, we can conclude that S. paradoxus
is evolving at a slower rate than S. cerevisiae or S. mikatae.Ā This could be due to generation time, but
also life cycle throughout the year.Ā
Wild-type species remain dormant most of the year in spores, until the
next blooming.Ā This causes fewer cell
divisions, hence fewer errors in replicating the DNA.
We can also observe differences in the rate of change of individual genes.Ā One case stands out as an extreme outlier: the mating-type gene MATA2.Ā The gene shows perfect 100% conservation at the amino acid level over its entire length (119 aa) across all four species. More strikingly, the gene shows perfect 100% conservation at the nucleotide level as well (357 bp). This differs sharply for the typical pattern seen for protein-coding genes, which show relaxed constraint in third positions of codons.Ā Notably, the MATA2 gene is the only one of the four mating-type genes (the others being MATa1, MATa2 and MATA1) whose biochemical function remains unknown despite two decades of research75. An important clue may be that the sequence of MATA2 is identical in all four species to the 3Æ-end of the MATa2 gene. Perfect conservation at the nucleotide-level and identity to the terminus of MATa2 suggests that MATA2 may function not only by encoding a protein, but by encoding an anti-sense RNA or a DNA site.Ā Hence, the lack of evolutionary change can suggest additional biological functions responsible for the pressure to conserve nucleotide sequence.
6.6. Evidence and mechanisms of rapid protein change. ĀĀĀ
Similarly, the unusually high rate of change can be biologically meaningful.Ā The gene analysis described in chapter 2 rejected only a single ORF (YBR184W) that is clearly encoding a functional protein.Ā The region containing YBR184W corresponds to a large open reading frame in all four species (524, 558, 554 and 556 amino acids, respectively), but the alignment shows unusually low sequence conservation. The sequence has only 32% nucleotide identity and 13% amino acid identity across the four species (Figure 6.4). Pairwise alignments across the species show numerous insertions and deletions, explaining why the gene failed the RFC test.Ā (Interestingly, multiple alignment of all four species simultaneously improves the alignment sufficiently that the gene passes the RFC test; this suggests a way to improve the test.)

The rapid divergence is suggestive of a gene under strong positive selection.
We tested this notion by calculating the Ka/Ks ratio (the normalized ratio of
amino-acid-altering substitutions to silent substitutions), a traditional test
for positive selection76. Whereas typical genes in S. cerevisiae show a Ka/Ks ratio of 0.11
▒ 0.02, YBR184W has a ratio of 0.689.
This ratio ranks as the third highest observed among all yeast genes (If three
small domains with high conservation are excluded, the ratio rises to
0.774).Ā The two genes with higher Ka/Ks
ratio are YAR068W, a putative
membrane protein, and YER121W, whose
expression changes under stress.Ā
The protein encoded by YBR184W has not been extensively studied, but expression studies show that the gene is induced during sporulation77 and sequence analysis shows that it is similar to the gene YSW1 that encodes a spore-specific protein. This is consistent with the observation that many of the best studied examples of positive selection in other organisms are genes related to gamete function.Ā The change might promote speciation by imposing constraints on mating partner selection.
The vast majority of nucleotide changes in protein coding regions are silent or affect individual amino acids.Ā However, a small number of events suggest additional mechanisms of rapid protein change.Ā These events include closely spaced compensatory indels that affect the translation of small contiguous amino acid stretches.Ā They also include the loss and gain of stop codons (by a nucleotide substitution or a frame-shifting indel) that may result in the rapid change of protein segments or the translation of previously non-coding regions78.Ā Such events are observed more frequently near telomeric regions and may affect silenced genes or recently inactivated pseudogenes.Ā
Additionally, we found a small number of differences in the length of orthologous proteins.Ā These typically involve changes in the copy number of tri-nucleotide repeats, such as (CAA)n that encodes hydrophobic stretches often involved in protein-protein interactions.Ā The most drastic example is seen for the TFP1 gene, which encodes a vacuolar ATPase.Ā The S. cerevisiae gene contains an insertion of 1400 bp that is absent in the three related species.Ā The insertion corresponds to the recent horizontal transfer of a known post-translationally self-splicing intein, VMA179.
6.7. Conclusion.
When comparing genomes, similarities and differences alike can reveal biological meaning.Ā In comparing closely related species, the precise ways in which genomes change can reveal important biological insights.Ā From the large-scale chromosomal changes, to the substitutions of individual nucleotides, we find specific rules and constraints in the ways genomes evolve.Ā Precise signals seem to govern how genomes are read, but also how they change.Ā Evolutionary fitness may come from the combination of a fit genome that outperforms competition in the present, but also a modular genome that enables rapid evolution in times of extreme environmental pressure.Ā The ability to rapidly carry out advantageous changes may be an inherent requirement in creating complexity via modularity.Ā Evolutionary traits may be selected by reversible changes that allowed survival in the past, and will allow survival in the future. Each of the similarities and differences observed merits further experimental study. Understanding how genomes are written, and how they change, will be central to our understanding of the ever-changing book of life.Ā