A Python based multi-paradigm simulation environment
It has always been the dream of scientists to predict the properties of new materials by theoretical modeling or computer simulation from a very fundamental, ab initio perspective. Within the coming decade, this dream will come true and we will be able to predict macroscopic properties of complex materials from first principles.
At the beginning of the new millennium, scientists are, with the advent of new very accurate quantum mechanical methods, enormous growth of computing power, and breakthroughs in computers sciences, for the first time able to foresee achieving this goal within a timeframe of the current decade.
The development of the Computational Materials Design Facility (CMDF) is a step towards the realization of this vision. The CMDF is developed in collaboration with the Materials and Process Simulation Center (MSC) at Caltech.
Additional information:
|
What is CMDF?
The CMDF is a new simulation framework that allows multi-scale multi-paradigm simulations of complex materials phenomena that operate on different length- and time scales. The method is based on a generic within a Python scripting environment, with the objective to enable complex multi-scale simulation tasks encompassing various simulation paradigms, such as quantum mechanics, reactive force fields, empirical force fields and continuum descriptions of materials. |
|
Different simulation modules are called from the scripting layer, which in turn call complex underlying routines. Interfaces between different codes, along with a central data structure allow straightforward communication between different simulation engines. We have been able to demonstrate that the method can be used to model materials phenomena such as crack propagation in silicon, oxidation of surfaces, or enzymatic processes in proteins. In particular, the combination of reactive potentials, along with classical non-reactive potentials is a very promising approach which can easily be achieved within the CMDF framework. Figure 1 summarizes the concept of overlapping scales and paradigms of a hierarchy of simulation methods to achieve coupling of QM scales up to the macro-engineering scale.
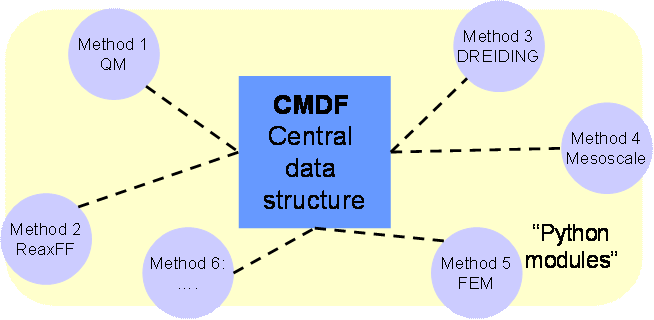
The CMDF is the first scale agnostic simulation method that enables integration of desperately different scales and paradigms in large-scale materials simulations. We expect that the CMDF will have significant scientific impact.
To which problems has CMDF been applied to? 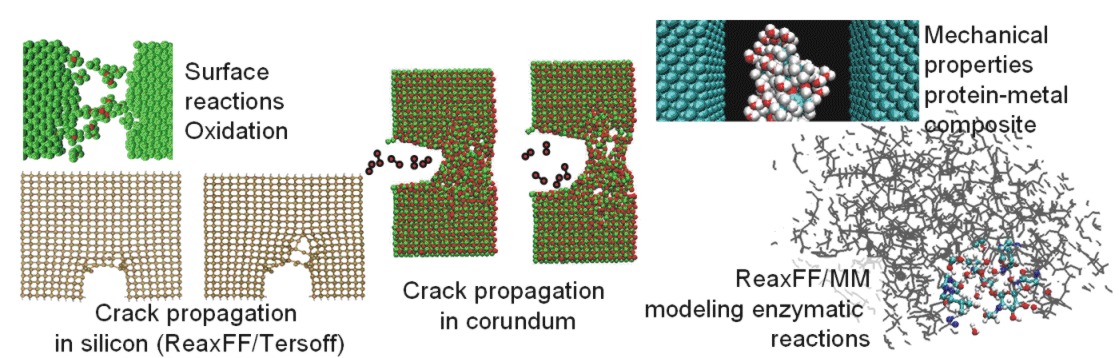
Applications of the CMDF framework
range from materials science (cracking in silicon), biology (enzymatic reactions), chemistry (oxidation) to novel hybrid bio-nano-materials. More information can be found in the CMDF related publications (see below) and in the proceedings of the 1st CMDF workshop.
Development of the CMDFat MIT's Atomistic Mechanics Modeling Laboratory is supported by the Army Research Office (ARO).
Publications
- M.J. Buehler , A.C.T. v. Duin, W.A. Goddard III, "Hybrid ReaxFF/Si-Tersoff modeling of dynamical crack propagation in silicon". Phys. Rev. Lett., March 10, pp. 095505, 2006 (link to paper...).
- M.J. Buehler , Jef Dodson, A.C.T. v. Duin, W.A. Goddard III, "The Computational Materials Design Facility (CMDF): A powerful framework for multiparadigm multi-scale simulations", Mat. Res. Soc. Proceedings (Combinatorial Methods and Informatics in Materials Science), Vol. 894, LL3.8, 2006 (link to paper...).
|
