




Solid Oxide Fuel Cells
High fuel flexibility of solid-oxide fuel cells (SOFCs) affords the possibility to use relatively cheap, safe, and readily available hydrocarbon (e.g., CH4) or coal syngas (i.e., H2-CO mixtures) fuels. Utilization of such fuels would greatly lower fuel cost and increase the feasibility of SOFC commercialization, especially for near-term adoption in anticipation of the long-awaited so-called "hydrogen economy." Current SOFC technology has shown good performance with a wide range of hydrocarbon and syngas fuels, but there are still significant challenges for practical application. Our research in this area is focused on creating detailed, multiscale, physics-based, computational models to facilitate better understanding of very specific elementary processes, to pinpoint rate-determining (and hence, performance-limiting) processes, and to provide performance predictions under a range of conditions.
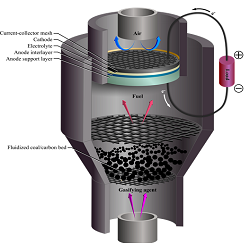
Direct Carbon Fuel Cells
A direct carbon fuel cell (DCFC) converts solid carbonaceous fuel, such as coal or biomass, directly into electricity with a fuel cell. One particularly promising DCFC design couples a fluidized bed coal gasifier with a SOFC, as shown in the conceptual drawing. This device is capable of attaining high thermodynamic efficiencies by thermally coupling the gasifier to the fuel cell. There are still many aspects of this coupling that are not well understood, such as the optimal temperature, geometry and flow-rate to enhance performance without wasting fuel. It is also currently unclear whether steam or carbon dioxide should be selected to flow through the gasifier. In order to explore these different options, an equilibrium gasifier model has been coupled to the 1D-MEA model to study the system’s performance. System efficiencies ranging from 50-60% are predicted for this system, making it a state-of-the-art alternative to traditional coal-fired power plants with only 35% efficiency.
Membrane Electrolyte Assembly (MEA) Model
The membrane-electrode assembly (MEA) is the heart of any fuel cell. In a SOFC, the MEA consists of a dense oxygen-ion-conducting electrolyte sandwiched between two porous electrodes. On the anode side, gas species in the fuel channel are transported through the porous cermet network of the electrode to electrochemically reactive sites known as triple- or three-phase boundaries (TPBs). It is generally accepted that current-producing charge-transfer reactions only occur at or very near the TPB, where the electron conductor (metal), ion conductor (oxide), and necessary gas-phase reactants (in the pore space) come together. Each phase is necessary for the transport of reactants to or removal of products from the reactive site. The interdependence of heterogeneous chemistry, charge-transfer reactions, and transport (ion, electron, and gas phase)---and the influence of electrode microstructure on these processes---determine the rate and ability of the cell to deliver current. We have developed, and continue to build on, a one-dimensional homogenized model of the MEA that is capable of incorporating multistep reaction mechanisms for homogeneous gas-phase chemistry and heterogeneous surface chemistry. Porous-media transport in the electrodes is represented with a dusty-gas model. Charge-transfer chemistry is represented in a modified Butler-Volmer formalism that requires a priori knowledge of a rate-limiting step (or the assumption of global charge transfer), but because this inherently assumes that charge transfer is the overall rate-limiting process, we are also developing elementary multistep charge-transfer mechanisms that are incorporated in the same way as the thermochemistry. Our models are built on a mathematical framework that can be applied to any MEA geometry.
Charge-Transfer Kinetics
Charge-transfer processes are among the least understood aspects of fuel cell chemistry. Computing the production rates from faradaic and nonfaradaic reactions requires the kinetic parameters for every reaction. In almost all cases, the charge-transfer kinetics are not known, and are instead used as fitting parameters to match model predictions with actual measured cell performance. Representing reactions in mass-action form does not suffer from restrictions that are inherent in the Butler-Volmer form. Only if the charge transfer is indeed rate limiting, such that the activities are established by much faster processes (e.g., adsorption/desorption), can the Butler-Volmer equation be used. To move away from a Butler-Volmer description of charge transfer, we are building a reaction database of single-electron-transfer reactions whose incorporation into patterned-anode and MEA models yields a good match to measured behavior of similar systems. This requires tuning the unknown electrochemical reaction kinetics so that model predictions are in best possible agreement with steady-state (Tafel) and time-variant (electrochemical impedance spectra, or EIS) responses. The multistep mechanism that is able to provide a good match to experimental data provides an understanding of the behavior at the TPB and the kinetics of that behavior.