1. Multiple Metal-Carbon Bonds
The goals of this research are to discover fundamentally new organometallic chemistry, new catalysts, and new principles of catalysis through guided exploratory synthesis and kinetic and mechanistic studies. The main type of organometallic chemistry that has evolved in the past 20 years has concerned complexes that contain a metal-carbon double bond (alkylidene complex) or triple bond (alkylidyne complex) in which the metal (often Mo, W, or Re) is in its highest possible oxidation state. The chemistry of high oxidation state transition metal alkyl complexes has been intimately linked to that of alkylidene compexes over the years, and is now the focus of research employing group 4 metals.
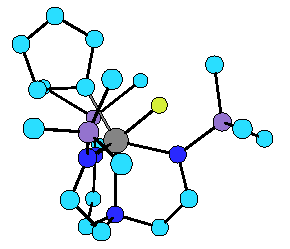
We have been involved in the synthesis and development of the organometallic chemistry of complexes that contain new multidentate multiamido ligands. One example are complexes that contain tetradentate triamidoamine (N3N) ligands, [(RNCH2CH2)3N]3- (R = Me3Si or C6F5), especially of the heavier, earlier transition metals (tantalum, molybdenum, tungsten, and rhenium). N3N ligands sterically protect ligands in the pocket formed by the three amido substituents and create a restricted orbital environment that contains two perpendicular pi type orbitals (dxz, dyz) and one sigma type (~dz2). Therefore metal-ligand double and triple bonds are highly favored. For example, we have found that addition of cyclopentyllithium to [N3N]WCl in ether yields the 18 electron cyclopentylidene hydride complex, [N3N]W(C5H8)(H), which appears to be the only documented example of a kinetically preferred alpha hydride elimination versus beta hydride elimination (see structure). In contrast,
X-ray structure of [(Me3SiNCH2CH2)3N]W(C5H8)(H).
[N3N]Mo(C5H9) is a stable, paramagnetic (high spin d2), purple crystalline compound. However, it has been shown that alpha elimination is also taking place at a rate of the order of the NMR time scale in this species, although the equilibrium lies toward the alkyl rather than the alkylidene hydride complex. In fact it has been shown that alpha elimination is taking place at a rate that is six to seven orders of magnitude faster than beta elimination, possibly because of steric hindrance within the trigonal pocket that encourages alpha elimination and hinders beta elimination. Both the Mo and W complexes decompose at greater than 50 degrees Centrigrade to give paramagnetic hydrides, [N3N]M(H). d2 Tungsten complexes of the type [(Me3SiNCH2CH2)3N]WCH2R are subject to "alpha,alpha-dehydrogenation" to give alkylidyne complexes, [(Me3SiNCH2CH2)3N]W(CR) and molecular hydrogen (R = H, Me, Pr, SiMe3, Ph, and CMe3). A cyclic alkyl such as cyclobutyl undergoes C-C bond cleavage and rearrangement to give a butylidyne complex.
Molybdenum (primarily) and W alkylidene complexes that are catalysts for the metathesis of carbon-carbon double bonds have been a focus of our research for several years. Molybdenum catalysts of the type Mo(CH-t-Bu)(NR')(OR)2 have been the subject of most research because they are relatively straightforward to prepare, they tolerate a variety of nonprotic functionalities, and their activity can be regulated through the choice of OR (e.g., from O-t-Bu to OCMe(CF3)2). Ring opening metathesis polymerization (ROMP) studies have included the consequences of reactivity differences and rates of interconversion of anti and syn rotamers of alkylidene complexes of the type Mo(CH-t-Bu)(N-2,6-R'2C6H3)(OR)2 in the ring-opening metathesis polymerization (ROMP) of norbornenes and substituted norbornadienes. Rotamer accessibility and relative reactivity will continue to be significant factors in metathesis reactions. In the last two years we have been focusing on the synthesis of catalysts that contain enantiomerically pure biphenolate or binaphtholate ligands in order to carry out asymmetric metathesis reactions that produce an enantiomerically pure product. An example would be the selective reaction of one enantiomer in a racemic mixture with an enantiomerically pure catalyst. The chiral Mo-alkylidene 1 contains the 5,5',6,6'-tetramethyl-3,3'-di-tert-butyl-1,1'-biphenyl-2,2'-diolate as the chiral ligand has been characterized through X-ray crystallography. Chiral Mo complex 1 effects enantioselective RCM (ring-closing metathesis) efficiently and with excellent selectivity. Depending on the substitution pattern of the reacting alkenes, either the diene substrate or the cycloalkenyl product can be obtained in 92% ee. More recent applications concern the "desymmetrization" of substrates having a plane of symmetry to give enantiomerically pure products in 95% yield and 95% ee. Examples of this and other recent results can be found in the schemes below. The ARCM (asymmetric RCM) technique promises to find wide application in organic chemistry since it delivers chiral products with high enantioselectivity and efficiency and can be carried in the absence of solvent and with low catalyst loadings.

Recent Relevant Papers: 294, 297, 298, 300, 303, 305, 314, 315, 316, 324, 326, 330, 334, 340, 346, 348.
High Oxidation State Dinitrogen Complexes
The ultimate goal of this project is to explore methods of activating and reducing dinitrogen using protons (or other electrophiles such as trimethylsilyl) and electrons, especially with V, Mo, W, and Re complexes in which the metal is in a relatively high oxidation state.
Recent efforts have been directed toward the synthesis of new ligands of the triamidoamine type and complexes thereof and an exploration of chemistry of V, Mo, W, and Re that is relevant to the activation of dinitrogen. For example, we recently reported that Mo[N3N'](OTf) ([N3N'] = [(C6F5NCH2CH2)3N]) can be reduced by one equivalent of sodium amalgam to give a paramagnetic dimeric dinitrogen complex, [N3N']Mo(mu-N2)Mo[N3N'], which can be further reduced by sodium amalgam to yield [N3N']Mo-N=N-NaLx (L = ether or THF). Recent results include the synthesis of molybdenum dinitrogen complexes that contain the TMStren ligand system, including the Mo(III) dinitrogen complex, [N3N]Mo(N2), and the Mo(II) species, {[N3N]Mo(N2)}- as its Mg2+ salt. An especially interesting development is the synthesis of a compound that contains three {[N3N]Mo(N2)}- ions around iron (see structure). This appears to be the first example of a dinitrogen complex that contains both iron and molybdenum. Since the most common nitrogenase enzyme contains seven iron atoms and one molybdenum atom, this structure may prove to be relevant to reduction of dinitrogen by that enzyme.

X-ray structure of {[(Me3SiNCH2CH2)3N]Mo(dinitrogen)}3Fe.
A significant fraction of the work at this stage is devoted to ligand design and synthesis. We hope to prepare ligands that are more resistant to cage cleavage reactions and reactions involving the amido substituents, and that therefore perhaps will be suitable for preparing complexes that will activate dinitrogen in a controlled and predictable manner. Recently we have developed reliable syntheses of triamidoamine complexes of Mo and W in which the triamidoamine ligand's substituents are ordinary aryl groups, e.g., p-t-butylphenyl. Architecturally more elaborate versions of such species we believe will allow the site for binding and reduction of dinitrogen to be relatively isolated, and in particular will prevent formation of bimetallic bridging dinitrogen complexes.
Recently we have found routes to Mo, W, and Re complexes that contain a diamidoamine ligand, [(RNCH2CH2)2NR']2- that we believe will also be of interest in terms of binding and reducing dinitrogen. These ligands too can be sterically and electronically varied to a significant degree, i.e., they can be tailored for the task required.
Recent Relevant Papers: 288, 296, 301, 302, 305, 306, 308, 312, 313, 314, 329, 332, 335, 337.
Controlled Polymerization of Alkenes by Catalytic Methods
For the last year we have been involved in the design and development of non-cyclopentadienyl catalysts for the polymerization of olefins in a living manner. The current focus is on "diamido/donor" ligands such as [(t-butylN-o-C6H4)2O]2- or the [t-BuNON]2- ligand. Such ligands employ four or five orbitals on the metal for bonding (three sigma, one or two pi), leaving five or four, respectively, for bonding to ligands. The most common types of complexes are trigonal bipyramidal complexes, for example of the type [t-BuNON]MR2 where M = Ti, Zr, or Hf and R is an alkyl ligand. In such species the donor ligand and one alkyl ligand occupy axial sites on the metal. The axial alkyl ligand can be removed using techniques analogous to those used to prepare polymerization catalysts based on group 4 metallocenes. An X-ray study of the [t-BuNON]ZrMe2complex activated with B(C6F5)3 is shown below.

X-ray structure of {[t-BuNON]ZrMe}{MeB(C6F5)3]
Activation of [t-BuNON]ZrMe2 with [PhNHMe2][B(C6F5)4] in chlorobenzene yields methane and an observable "cationic" catalyst that will polymerize 500 equivalents of 1-hexene to give atactic poly(1-hexene) with a molecular weight and polydispersity (1.03-1.10) characteristic of a living polymerization process. To our knowledge this is the first time that the chain propagating species in a group 4 metal catalyzed addition polymerization process has been observed. As a consequence of these results, addition polymerization of olefins by group 4 metal catalysts has become the major focus of this research program. The potential utility of a variety of diamido/donor ligands in early transition metal chemistry for a variety of purposes is also being examined closely.
We have recently begun to prepare a variety of "diamido/donor" ligand group 4 metal complexes. Compounds of the type (ArylNHCH2CH2)2O (Aryl = 2,6-Me2C6H3 (H21a), 2,6-Et2C6H3 (H21b), or 2,6-i-Pr2C6H3 (H21c)) can be prepared by treating (TsOCH2CH2)2O (TsO = tosylate) with the lithium anilides in THF. [1a,b]TiCl2, [1a,b]TiMe2, [1a]Ti(CH2Ph)2, [1a-c]M(NMe2)2 (M = Zr or Hf), [1a-c]MCl2, and [1a-c]MR2 (R = Me, Et, i-Bu) were prepared. An X-ray study of [1a]Ti(CH2Ph)2 revealed the structure to be a distorted trigonal bipyramid (type B) in which the two amido nitrogens and one benzyl ligand occupy equatorial positions. An X-ray study of [1a]ZrMe2 showed it to be a distorted trigonal bipyramid that contains "axial" amido groups (type A), while an X-ray study of [1c]HfEt2 revealed it to have a structure half-way between type A and type B, i.e., a distorted square pyramid with one alkyl in the apical position (see below). Addition of one equivalent of [PhNMe2H][B(C6F5)4] in C6D5X (X = Br, Cl) to [1a,c]MMe2 (M = Zr, Hf) gave cationic complexes that contain coordinated dimethylaniline, with which free aniline does not exchange readily on the NMR time scale.

X-ray structure of [(2,6-i-Pr2C6H3NCH2CH2)2O]HfEt2
Addition of excess ether to {[1a]MMe(NMe2Ph)}[B(C6F5)4] (M = Zr, Hf) led to {[1a]MMe(ether)}[B(C6F5)4] (M = Zr, Hf) complexes in high yield. Zr and Hf dimethyl complexes can be activated with [Ph3C][B(C6F5)4] to yield efficient catalysts for polymerization of 1-hexene; the molecular weight of the poly(1-hexene) chains is limited to ~20,000 to ~25,000 under the conditions employed. Neither {[1c]ZrMe(ether)}[B(C6F5)4] nor {[1c]HfMe(ether)}[B(C6F5)4] will polymerize 1-hexene in C6D5Br at room temperature, and neither will polymerize ethylene readily at one atmosphere and room temperature. It is proposed that a five-coordinate cation must lose a base in order to react with an olefin and that unimolecular chain termination (e.g., beta hydride elimination) in the four-coordinate cation limits chain length.
Recent Relevant Papers: 290, 293, 295, 304, 309, 319, 328, 331, 338, 339, 341, 342, 343, 344.
![]() This web page created by Eric Schrock, copyright 1995.
This web page created by Eric Schrock, copyright 1995.
All material within is the property of Richard Schrock. All Rights Reserved.
Any changes, updates, etc. that are required please email rrs@mit.edu