The XSF format is internal
XCrySDen structure
format. XSF stands for
XCrySDen Structure
File. It is used to describe (i) molecular and
crystal structure, (ii) forces acting on constituent
atoms, and (iii) scalar fields (for example: charge
density, electrostatic potential). The main attributes
of XSF format are:
- all records are in free format
- the XSF formatted file is composed from various
sections
- each sections begins with the keyword
- there are two types of keywords: (i)
single keywords, and (ii) sandwich
keywords, which are defined as:
-
single keyword: section begins with a
single keyword and ends without an end-keyword
-
sandwich keyword: section begins with
a begin-keyword (i.e.
BEGIN_keyword) and ends with an
end-keyword (i.e. END_keyword),
where keyword is one among
keywords.
- all coordinates are in ANGSTROMS units
- all forces are in Hartree/ANGSTROM units
- the comment-lines start with the "#" character
(see below)
The comment-lines were introduced to the XSF file
starting from
XCrySDen version 1.4.
The comment-lines start with the
"#" character;
comments are allowed only between sections and not
within a given section. Here are two examples (correct
and incorrect).
Correct example (comments are bewteen the
sections):
# this is a specification
# of ZnS crystal structure
CRYSTAL
# these are primitive lattice vectors (in Angstroms)
PRIMVEC
2.7100000000 2.7100000000 0.0000000000
2.7100000000 0.0000000000 2.7100000000
0.0000000000 2.7100000000 2.7100000000
# these are convetional lattice vectors (in Angstroms)
CONVVEC
5.4200000000 0.0000000000 0.0000000000
0.0000000000 5.4200000000 0.0000000000
0.0000000000 0.0000000000 5.4200000000
# these are atomic coordinates in a primitive unit cell
# (in Angstroms)
PRIMCOORD
2 1
16 0.0000000000 0.0000000000 0.0000000000
30 1.3550000000 -1.3550000000 -1.3550000000
|
Incorrect example (comment is
within the section):
ATOMS
6 2.325243 -0.115261 0.031711
1 2.344577 -0.363301 1.077589
#
# this is a comment on the wrong place
#
6 0.007719 -0.041269 0.244204
9 0.064656 1.154700 0.824420
9 -0.042641 -0.911850 1.255074
8 -1.071578 -0.152842 -0.539134
|
For MOLECULES the XSF format is very simple. The
first line begins with the
ATOMS keyword
and then one specifies the structural data for all
atoms. An entry for an atom looks like:
AtNum X Y Z
where
AtNum stands for atomic number (or symbol), while
X Y Z are Cartesian coordinates in ANGSTROMS units. Here is one
example:
ATOMS
6 2.325243 -0.115261 0.031711
1 2.344577 -0.363301 1.077589
9 3.131708 -0.909527 -0.638930
9 2.736189 1.130568 -0.134093
8 1.079338 -0.265162 -0.526351
6 0.007719 -0.041269 0.244204
9 0.064656 1.154700 0.824420
9 -0.042641 -0.911850 1.255074
8 -1.071578 -0.152842 -0.539134
6 -2.310374 0.036537 0.022189
1 -2.267004 0.230694 1.077874
9 -2.890949 1.048938 -0.593940
9 -3.029540 -1.046542 -0.203665
|
The XSF format allows to specify structures of
different periodicity. The keywords
MOLECULE,
POLYMER,
SLAB, and
CRYSTAL designate
the 0-, 1-, 2-, and 3-dimensional structures (i.e.
periodic dimensions are meant here). For crystal
structures the file begin with
CRYSTAL
keyword. Then one needs to specify the lattice vectors
and the atoms belonging to the unit cell.
XCrySDen accepts two
different setting of the unit cell. These are usually
called the primitive and the conventional unit cell.
The corresponding keywords are
PRIMVEC for
primitive lattice vectors and
PRIMCOORD
for atoms belonging to the primitive unit cell. The
CONVVEC and
CONVCOORD have
the analogous meaning for the conventional unit cell.
Warning: in XSF file the two settings of the
unit cell are supposed to have the same origin. If you
want to specify some crystal structure with two
settings of the unit cell, that have different origins
you will have to create two XSF files. In practice only
PRIMVEC,
PRIMCOORD and
CONVVEC need to be specified, and then
"conventional coordinates" (
CONVCOORD) are
generated by
XCrySDen itself, hence
it is
never needed to specify the
CONVCOORD section !!! Here is an example
of ZnS crystal structure:
CRYSTAL see (1)
PRIMVEC
0.0000000 2.7100000 2.7100000 see (2)
2.7100000 0.0000000 2.7100000
2.7100000 2.7100000 0.0000000
CONVVEC
5.4200000 0.0000000 0.0000000 see (3)
0.0000000 5.4200000 0.0000000
0.0000000 0.0000000 5.4200000
PRIMCOORD
2 1 see (4)
16 0.0000000 0.0000000 0.0000000 see (5)
30 1.3550000 -1.3550000 -1.3550000
|
|
Legend:
|
|
(1)
|
this keyword specify the structure is crystal
|
|
(2)
|
specification of PRIMVEC (in
ANGSTROMS) like:
ax, ay, az (first
lattice vector)
bx, by, bz (second
lattice vector)
cx, cy, cz (third
lattice vector)
|
|
(3)
|
specification of CONVVEC (see
(2))
|
|
(4)
|
First number stands for number of atoms in
the primitive cell (2 in this case). The
second number is always 1 for
PRIMCOORD coordinates.
|
|
(5)
|
Specification of atoms in the primitive cell
(the same as for ATOMS section).
|
All that is needed to specify the forces acting on
atoms is to supplement the appropriate coordinate
section (
ATOMS or
PRIMCOORD).
Now an entry for an atom would look like:
AtNum X Y Z Fx Fy Fz
where
Fx Fy Fz stands for force components in X, Y and Z
direction, respectively. The force components are expressed in Cartesian
coordinate system in Hartree/ANGSTROM unit. Here is an example of water
molecule:
ATOMS
8 0.00000 0.00000 0.00000 -.05164 .00000 -.03999
1 0.00000 0.00000 1.00000 .01769 .00000 .03049
1 0.96814 0.00000 -0.25038 .03395 .00000 .00949
|
And here is an example for the periodic
structure structure:
SLAB
PRIMVEC
5.8859828533 0.0000000000 0.0000000000
0.0000000000 5.8859828533 0.0000000000
0.0000000000 0.0000000000 1.0000000000
PRIMCOORD 1
11 1
6 3.674759 2.942992 -3.493103 -0.021668 0.000000 -0.057324
1 4.121990 3.816734 -4.007689 -0.000478 0.001204 0.006657
1 4.121990 2.069250 -4.007689 -0.000478 -0.001204 0.006657
6 2.211226 2.942992 -3.493103 0.021668 0.000000 -0.057324
1 1.763995 3.816734 -4.007689 0.000478 0.001204 0.006657
1 1.763995 2.069250 -4.007689 0.000478 -0.001204 0.006657
8 0.000000 0.000000 -2.719012 0.000000 0.000000 -0.050242
47 4.448147 4.449892 -1.919011 -0.022812 -0.029123 0.007553
47 4.448147 1.436093 -1.919011 -0.022812 0.029123 0.007553
47 1.437838 4.449892 -1.919011 0.022812 -0.029123 0.007553
47 1.437838 1.436093 -1.919011 0.022812 0.029123 0.007553
|
It is possible to specify several snapshots of a
structure in the XSF format. For example: the file can
contain the data for all structures that were created
during an optimization run or molecular-dynamics run.
These structures can be displayed as animation by
XCrySDen.
The main attributes of the animated XSF format are:
- the AXSF file begins with the
ANIMSTEPS
nstep keyword, where
nstep stands for number of
animation steps.
- number of atoms for all steps must be the
same !!!
- in fixed-cell animated XSF for crystal structures
the unit cell for all steps is taken to be the same
- in variable-cell animated XSF for crystal
structures the unit cell is specified for each
animation step
- the
ATOMS and PRIMCOORD
keywords are prefixed by an integer number which is
the sequential index of a given snapshot. Therefore
the syntax is ATOMS istep and
PRIMCOORD istep.
- for variable-cell crystal structure the
PRIMVEC and CONVVEC
keywords are prefixed with sequential index.
Here is an example of AXSF file. It shows
different structures during an optimization of water
molecule. Note the index prefixes after
ATOMS keywords.
ANIMSTEPS 4
ATOMS 1
8 0.0000 0.0000 0.0000 -0.0516 0.0000 -0.0399
1 0.0000 0.0000 1.0000 0.0176 0.0000 0.0304
1 0.9681 0.0000 -0.2503 0.0339 0.0000 0.0094
ATOMS 2
8 -0.1480 0.0000 -0.1146 0.0020 0.0000 0.0015
1 -0.0468 0.0000 0.9134 -0.0069 0.0000 0.0069
1 0.8726 0.0000 -0.2740 0.0049 0.0000 -0.0084
ATOMS 3
8 -0.1032 0.0000 -0.0799 0.0013 0.0000 0.0010
1 -0.0319 0.0000 0.9591 0.0011 0.0000 -0.0028
1 0.9205 0.0000 -0.2710 -0.0025 0.0000 0.0018
ATOMS 4
8 -0.1102 0.0000 -0.0853 0.0001 0.0000 0.0000
1 -0.0345 0.0000 0.9503 -0.0000 0.0000 -0.0000
1 0.9114 0.0000 -0.2714 -0.0000 0.0000 -0.0000
|
Here is an example of animated XSF for the ZnS
crystal structure with the fixed unit-cell. Note the
index prefixes after
PRIMCOORD keywords.
ANIMSTEPS 2
CRYSTAL
PRIMVEC
0.0000000 2.7100000 2.7100000
2.7100000 0.0000000 2.7100000
2.7100000 2.7100000 0.0000000
PRIMCOORD 1
2 1
16 0.0000000 0.0000000 0.0000000
30 1.3550000 -1.3550000 -1.3550000
PRIMCOORD 2
2 1
16 0.0000000 0.0000000 0.0000000
30 1.2550000 -1.2550000 -1.2550000
|
Here is an example of animated XSF for the ZnS
crystal structure with the variable unit-cell. Note the
index prefixes after
PRIMVEC,
CONVVEC, and
PRIMCOORD
keywords.
ANIMSTEPS 2
CRYSTAL
PRIMVEC 1
2.7100000 2.7100000 0.00000000
2.7100000 0.0000000 2.71000000
0.0000000 2.7100000 2.71000000
CONVVEC 1
5.4200000 0.0000000 0.00000000
0.0000000 5.4200000 0.00000000
0.0000000 0.0000000 5.42000000
PRIMCOORD 1
2 1
16 0.0000000 0.0000000 0.00000000
30 1.3550000 -1.3550000 -1.35500000
PRIMVEC 2
2.9810000 2.9810000 0.00000000
2.9810000 0.0000000 2.98100000
0.0000000 2.9810000 2.98100000
CONVVEC 2
5.9620000 0.0000000 0.00000000
0.0000000 5.9620000 0.00000000
0.0000000 0.0000000 5.96200000
PRIMCOORD 2
2 1
16 0.0000000 0.0000000 0.00000000
30 1.5905000 -1.5905000 -1.59050000
|
It is possible to specify one or several scalar
fields (both 2D and 3D) as an
uniform mesh of
values in the XSF formatted file. The mesh can be
non-orthogonal. This scalar field meshes are
called
datagrids.
Datagrids in
XCrySDen are general
grids and not periodic one!!! What is meant by this
will be explained bellow. A general grid is a uniform
grid that is spanned inside some box, which is either
non-orthogonal or orthogonal. For a molecule such a box
would be a bounding box, while for crystal such a box
would be a unit cell. However in a grid that span a
whole unit-cell some border points would be redundant
due to translational symmetry. Grids that omit these
redundant points are called periodic (by this
convention a FFT (fast-Fourier transform) mesh would be
a periodic mesh). The general vs. periodic grid concept
is shown graphically here:
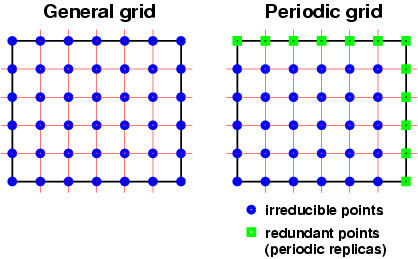
Note: datagrids in
XCrySDen are general
grids !!!
The datagrid section is structured and organized
in blocks. The grids inside a block can be manipulated
among themselves by
XCrySDen program. The
main attributes of XSF datagrids are:
|
(1)
|
the top datagrid section is called
BLOCK_DATAGRID and is sandwiched between
BEGIN_BLOCK_DATAGRID_xD and
END_BLOCK_DATAGRID_xD keywords,
where x stands for the
dimensionality of the grid. Currently 2D and
3D scalar grids are supported.
|
|
(2)
|
there can be an arbitrary number of
BLOCK_DATAGRID sections.
|
|
(3)
|
there can be an arbitrary number of datagrids
inside one BLOCK_DATAGRID section. All
datagrids belonging to the same
BLOCK_DATAGRID must have the dimensionality
as defined by
BEGIN_BLOCK_DATAGRID_xD keyword.
These datagrids must also share the following
properties: (i) they should span the same
space (origin and the spanning vectors must
be the same), and (ii) they should have the
same number of data-points. Each datagrid in
a BLOCK_DATAGRID is sandwiched inside
BEGIN_DATAGRID_xD_identifier and
END_DATAGRID_xD keywords, where
x stands for dimensionality of
the grid and the identifier is
used as an identifier of the datagrid.
|
|
(4)
|
after the
BEGIN_BLOCK_DATAGRID_xD keyword
and before the first
DATAGRID_xD_identifier keyword
is a comment, which is used as an identifier
for the BLOCK_DATAGRID. It must be a
single word!!!
|
|
(5)
|
the values inside a datagrid are specified in
column-major (i.e. FORTRAN) order. This means
that values are written as:
- C-syntax:
for (k=0; i<nz; k++)
for (j=0; j<ny; j++)
for (i=0; k<nx; i++)
printf("%f",value[i][j][k]);
- FORTRAN syntax:
write(*,*)
$ (((value(ix,iy,iz),ix=1,nx),iy=1,ny),iz=1,nz)
|
Above specifications may sound very fuzzy,
hence let us clarify this by looking at a very simple
example. The
description of all records
follows after the example. Also take a look at
schematic presentation of the
structure of datagrids of below example.
l.01
l.02
l.03
l.04
l.05
l.06
l.07
l.08
l.09
l.10
l.11
l.12
l.13
l.14
l.15
l.16
l.17
l.18
l.19
l.20
l.21
l.22
l.23
l.24
l.25
l.26
l.27
l.28
l.29
l.30
l.31
l.32
l.33
l.34
l.35
l.36
l.37
l.38
l.39
l.40
l.41
l.42
l.43
l.44
l.45
l.46
l.47
l.48
l.49
l.50
l.51
l.52
l.53
l.54
l.55
l.56
l.57
l.58
l.59
l.60
l.61
l.62
l.63
l.64
l.65
|
BEGIN_BLOCK_DATAGRID_2D
my_first_example_of_2D_datagrid
BEGIN_DATAGRID_2D_this_is_2Dgrid#1
5 5
0.0 0.0 0.0
1.0 0.0 0.0
0.0 1.0 0.0
0.000 1.000 2.000 5.196 8.000
1.000 1.414 2.236 5.292 8.062
2.000 2.236 2.828 5.568 8.246
3.000 3.162 3.606 6.000 8.544
4.000 4.123 4.472 6.557 8.944
END_DATAGRID_2D
BEGIN_DATAGRID_2D_this_is_2Dgrid#2
5 5
0.0 0.0 0.0
1.0 0.0 0.0
0.0 1.0 0.0
4.000 4.123 4.472 6.557 8.944
3.000 3.162 3.606 6.000 8.544
2.000 2.236 2.828 5.568 8.246
1.000 1.414 2.236 5.292 8.062
0.000 1.000 2.000 5.196 8.000
END_DATAGRID_2D
END_BLOCK_DATAGRID_2D
BEGIN_BLOCK_DATAGRID_3D
my_first_example_of_3D_datagrid
BEGIN_DATAGRID_3D_this_is_3Dgrid#1
5 5 5
0.0 0.0 0.0
1.0 0.0 0.0
0.0 1.0 0.0
0.0 0.0 1.0
0.000 1.000 2.000 5.196 8.000
1.000 1.414 2.236 5.292 8.062
2.000 2.236 2.828 5.568 8.246
3.000 3.162 3.606 6.000 8.544
4.000 4.123 4.472 6.557 8.944
1.000 1.414 2.236 5.292 8.062
1.414 1.732 2.449 5.385 8.124
2.236 2.449 3.000 5.657 8.307
3.162 3.317 3.742 6.083 8.602
4.123 4.243 4.583 6.633 9.000
2.000 2.236 2.828 5.568 8.246
2.236 2.449 3.000 5.657 8.307
2.828 3.000 3.464 5.916 8.485
3.606 3.742 4.123 6.325 8.775
4.472 4.583 4.899 6.856 9.165
3.000 3.162 3.606 6.000 8.544
3.162 3.317 3.742 6.083 8.602
3.606 3.742 4.123 6.325 8.775
4.243 4.359 4.690 6.708 9.055
5.000 5.099 5.385 7.211 9.434
4.000 4.123 4.472 6.557 8.944
4.123 4.243 4.583 6.633 9.000
4.472 4.583 4.899 6.856 9.165
5.000 5.099 5.385 7.211 9.434
5.657 5.745 6.000 7.681 9.798
END_DATAGRID_3D
END_BLOCK_DATAGRID_3D
|
|
Legend:
|
|
l.01
|
beginning of 2D block of datagrids
|
|
l.02
|
one word comment - used as an identifier for
this block
|
|
l.03
|
beginning of the first 2D datagrid in a block
|
|
l.04
|
number of data-points in each direction (i.e.
nx ny for 2D grids)
|
|
l.05
|
origin of the datagrid
|
|
l.06
|
first spanning vector of the datagrid
|
|
l.07
|
second spanning vector
|
|
l.08-12
|
5x5 datagrid values in column-major mode
|
|
l.13
|
end of the first 2D datagrid in a block
|
|
l.14-24
|
these lines specify second 2D datagrid in a
block in totally analogous manner as the
first 2D datagrid.
|
|
l.25
|
end of 2D block of datagrids
|
|
|
|
|
l.27
|
beginning of 3D block of datagrids
|
|
l.28
|
one word comment - used as an identifier for
this block
|
|
l.29
|
beginning of the first 3D datagrid in a block
|
|
l.30
|
number of data-points in each direction (i.e.
nx ny nz for 3D grids)
|
|
l.31
|
origin of the datagrid
|
|
l.32
|
first spanning vector of the datagrid
|
|
l.33
|
second spanning vector
|
|
l.34
|
third spanning vector
|
|
l.35-63
|
5x5x5 datagrid values in column-major mode
|
|
l.64
|
end of the first 3D datagrid in a block
|
|
l.65
|
end of 3D block of datagrids
|
Here is a scheme of the structure of datagrids
of above example as displayed by
XCrySDen program.
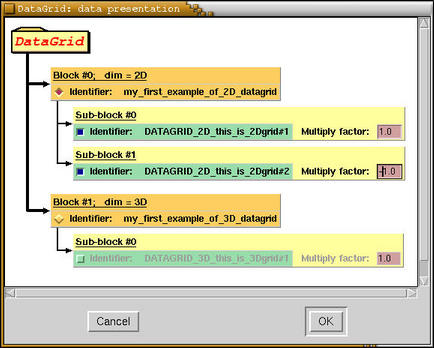
Bandgrids are variation on a theme of
datagrids. They were introduced for the visualization
of Fermi surfaces and yields a simplified format of
datagrids. Please notice that despite their
purpose they are still
general
grids and not periodic ones. The
bandgrid
for the visualization of Fermi surface should be
defined as:
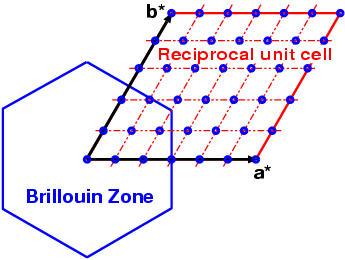
Please notice that the bandgrid span the
reciprocal unit cell, not the Brillouin zone !!!
Points are later translated back to Brillouin zone by
XCrySDen
and Fermi surface is rendered inside the Brillouin
zone.
Warning: so far
XCrySDen uses
bandgrids solely for the visualization of
Fermi surfaces (the coresponding files are called
BXSF, standing for Band-XSF). If you want to render
some other property as contours/isosurfaces you must
specify the scalar fields as
datagrids. This
is because the Fermi surfaces needs some special
processing and therefore bandgrids are automatically
processed. The main attributes of
bandgrids
are:
|
(1)
|
the structure of bandgrids is
similar to datagrids
|
|
(2)
|
there can only be one BLOCK_BANDGRID section
|
|
(3)
|
there can only be one BANDGRID in
BLOCK_BANDGRID
|
|
(4)
|
there can be an arbitrary number of
bands inside BANDGRID section, where
BAND is ...
|
|
(5)
|
so far only 3D bandgrids are supported
|
|
BEWARE:
|
the values inside a bandgrid are specified in
row-major
(i.e. C) order. This means that values are
written as:
- C-syntax:
for (i=0; i<nx; i++)
for (j=0; j<ny; j++)
for (k=0; k<nz; k++)
printf("%f",value[i][j][k]);
- FORTRAN syntax:
write(*,*)
$ (((value(ix,iy,iz),iz=1,nz),iy=1,ny),ix=1,nx)
|
Here is a
bandgrid example. The
description of the records is below the example.
l.01
l.02
l.03
l.04
l.05
l.06
l.07
l.08
l.09
l.10
l.11
l.12
l.13
l.14
l.15
l.16
l.17
l.18
l.19
l.20
l.21
l.22
l.23
l.24
l.25
l.26
l.27
l.28
l.29
l.30
l.31
l.32
l.33
l.34
l.35
l.36
l.37
l.38
l.39
l.40
l.41
l.42
l.43
l.44
l.45
l.46
l.47
l.48
l.49
l.50
l.51
l.52
l.53
l.54
l.55
l.56
l.57
l.58
l.59
l.60
l.61
l.62
l.63
|
BEGIN_INFO
#
# this is a sample Band-XCRYSDEN-Structure-File
# aimed for Visualization of Fermi Surface
#
# Case: just an example
#
# Launch as: xcrysden --bxsf example.bxsf
#
Fermi Energy: 0.83511
END_INFO
BEGIN_BLOCK_BANDGRID_3D
here_we_have_some_examples
BEGIN_BANDGRID_3D_simple_example
2
4 4 4
0.0 0.0 0.0
1.0 0.0 0.0
0.0 1.0 0.0
0.0 0.0 1.0
BAND: 3
0.000 0.192 0.385 0.577
0.192 0.272 0.430 0.609
0.385 0.430 0.544 0.694
0.577 0.609 0.694 0.816
0.192 0.272 0.430 0.609
0.272 0.333 0.471 0.638
0.430 0.471 0.577 0.720
0.609 0.638 0.720 0.839
0.385 0.430 0.544 0.694
0.430 0.471 0.577 0.720
0.544 0.577 0.667 0.793
0.694 0.720 0.793 0.903
0.577 0.609 0.694 0.816
0.609 0.638 0.720 0.839
0.694 0.720 0.793 0.903
0.816 0.839 0.903 1.000
BAND: 4
1.000 0.942 0.885 0.827
0.942 0.918 0.871 0.817
0.885 0.871 0.837 0.792
0.827 0.817 0.792 0.755
0.942 0.918 0.871 0.817
0.918 0.900 0.859 0.809
0.871 0.859 0.827 0.784
0.817 0.809 0.784 0.748
0.885 0.871 0.837 0.792
0.871 0.859 0.827 0.784
0.837 0.827 0.800 0.762
0.792 0.784 0.762 0.729
0.827 0.817 0.792 0.755
0.817 0.809 0.784 0.748
0.792 0.784 0.762 0.729
0.755 0.748 0.729 0.700
END_BANDGRID_3D
END_BLOCK_BANDGRID_3D
|
|
Legend:
|
|
l.01
|
beginning of INFO section
|
|
l.02-09
|
a hash "#" character stands for comment
|
|
l.10
|
a "Fermi Energy:" string is a
marker for reading the value of Fermi energy
|
|
l.11
|
end of INFO section
|
|
|
|
|
l.13
|
beginning of 3D block of bandgrids
|
|
l.14
|
one word comment - used as an identifier for
this block
|
|
l.15
|
beginning of the 3D bandgrid
|
|
l.16
|
number of bands in the bandgrid
|
|
l.17
|
number of data-points in each direction (i.e.
nx ny nz for 3D grids)
|
|
l.18
|
origin of the bandgrid
Warning: origin should be (0,0,0)
(i.e. Gamma point)
|
|
l.19
|
first spanning vector of the bandgrid (i.e.
first reciprocal lattice vector)
|
|
l.20
|
second spanning vector
|
|
l.21
|
third spanning vector
|
|
l.22
|
beginning of the BAND with label 3
|
|
l.23-41
|
4x4x4 values in row-major mode
|
|
l.42
|
beginning of the BAND with label 4
|
|
l.43-61
|
4x4x4 values in row-major mode
|
|
l.62
|
end of the bandgrid
|
|
l.63
|
end of 3D bandgrid block
|








![[Figure]](img/xcrysden-picture-small-new.jpg)