It is possible to use
XCrySDen as a tool when
performing the analysis of computed properties.
XCrySDen
provides a subset of options among all the options of
the CRYSTAL
properties program. This chapter
is meant as a hand-on tutorial and we will go trough
all options that are available in
XCrySDen. First we
should compute some sample case (for examples one among
the test cases that come with standard CRYSTAL
distributions). Then we do something like:
integrals < testCase.d12 > testCase.out
scf > testCase.out
When the
testCase is finished
successfully, among other scratch files also the
fortran unit 9 is produced (usually is named as
fort.9 or
ftn9 depending on
the compiler). Then I usually do something like:
cp fort.9 testCase.f9 Now we can load this
file into
XCrySDen. This can be
achieved via two ways:
|
(1)
|
via command line option, i.e: xcrysden
--crystal_f9 testCase.f9
|
|
(2)
|
specifying a filename of unit 9 via
File-->Open
CRYSTAL-95/98/03 Properties
menu
|
After we load our unit 9 into
XCrySDen, the program
displays the structure and the
Properties menubutton gets
activated. All
properties options of
XCrySDen are
available under the
properties menubutton.
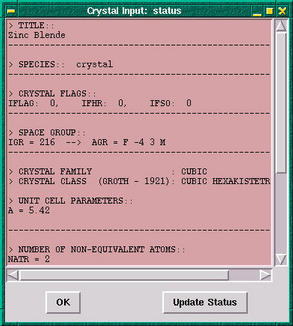
The first option in the
properties menu is the
Get INFO
option. This option prints the basic information about
the case. After selecting this option the program will
ask whether we want the band widths to be included in
the basic info. If we answer positively, then the the
following window appears:
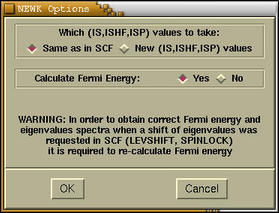
At the top of the
NEWK Options
window we can enter new NEWK values (refer to
CRYSTAL95/98 User Manual for the explanation
of the NEWK keyword). Below we can select whether we
want to recompute the Fermi energy. Answer according to
your needs. After pressing the
[OK] button a new
window appears, which holds a CRYSTAL output data.
This is a very useful option, which is accessible
under the
Display
Bandwidths menu. First the
NEWK window pops-up, where one can
select new NEWK values (refer to
CRYSTAL95/98 User
Manual for the explanation of the NEWK keyword)
and requests for the recalculation of the Fermi energy.
When we answer according to our needs the following
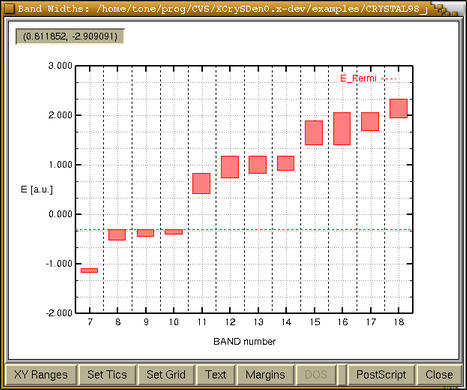
bar-graph window with band widths appears:
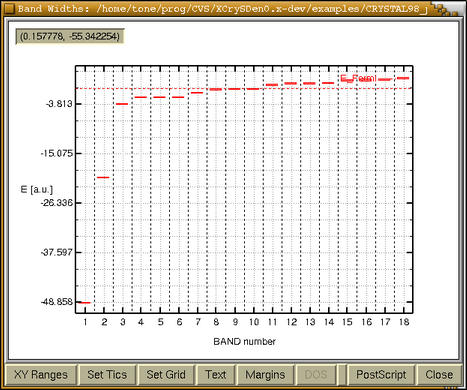
We immediately recognized that bands 1 and
2 are core states, bands from 3 to 6 are semi-core
states, bands from 7-10 are valence bands, whereas
bands from 11 on are unoccupied. Now we can refine our
plot pressing the
[XY Ranges] button and after
playing a while with other buttons, we get:
Density of States (DOS) can be calculated and
displayed via
Density of
States menu. First the
NEWK window pops-up, where one can
select new NEWK values (refer to
CRYSTAL95/98 User
Manual for the explanation of the NEWK keyword)
and requests for the recalculation of the Fermi energy.
When we answer according to our needs a new window
appears, where we specify the DOS parameter:
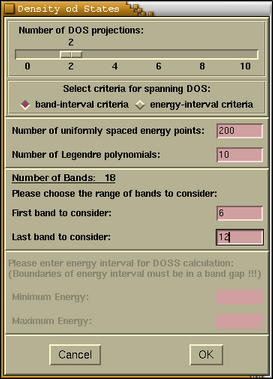
If we set the
number of DOS
projections (NPRO) to zero (default), then solely
total DOS will be computed. For the explanation of
other parameters refer to
CRYSTAL95/98 User
Manual. When we input all parameters, then if the
NPRO is zero the total DOS and another window holding
CRYSTAL output appear. On the other hand, if the NPRO
is greater then zero, then a new window appears. Here
we enter the projection parameters for each projection.
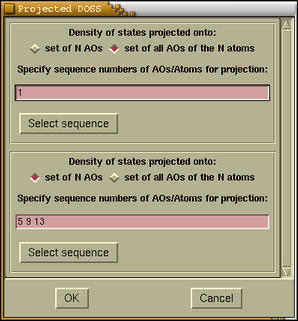
It is possible either to project the DOS
to
set of some atomic orbitals (AOs) or to
set of all AOs for some group of atoms. This
can be determining by choosing correct radiobutton. If
we select the first possibility then we should enter
the
ID numbers of appropriate AOs. In CRYSTAL
AOs are numbered in sequential fashion. To get an
ID number for a particular AO we must know:
(i) the order of atoms, (ii) the basis set, and (iii)
how the shell are ordered in CRYSTAL (the order for
p type shell is:
x, y, z, while order
for
d type shell is:
2z2-x2-y2, xz, yz,
x2-y2, xy; refer to
CRYSTAL95/98 User Manual, p.50) Some
information, such as number of AOs, and sequential
order of atoms can be obtained by reading the output
provided by
Properties-->Get INFO menu
option. If we press the
[Select sequence]
button, then a selection window appears. It merely
displays the ID numbers of either AOs or atoms
(depending on the prior choice of projection-type).
Here we can select appropriate numbers by holding down
the
CTRL key and mouse-clicking.

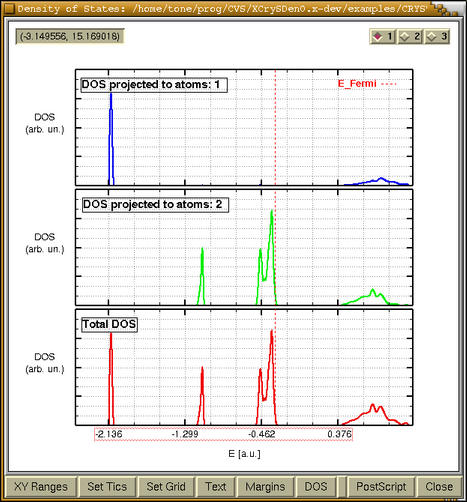
When we are done then after pressing the
[OK] button the projected DOS (only if it was
specified) and total DOS appear. Also another window
holding the CRYSTAL output pops-up. Via
[XY
Ranges],
[Set Tics],
[Set Grid],
[Text],
[Margins] and
[Dos]
buttons one can refine the plot and make its appearance
nicer. If one wants to change the font for labels or
width/color of curves one can simple double click these
items on the plot and a
configuration window
will pop-up.
Band Structure (BAND) can be calculated and
displayed via
Band
Structure menu. First the
NEWK window pops-up, where one can
select new NEWK values (refer to
CRYSTAL95/98 User
Manual for the explanation of the NEWK keyword)
and requests for the recalculation of the Fermi energy.
When we answer according to our needs a new window
appears, where we select a k-path inside Brillouin zone
(BZ).
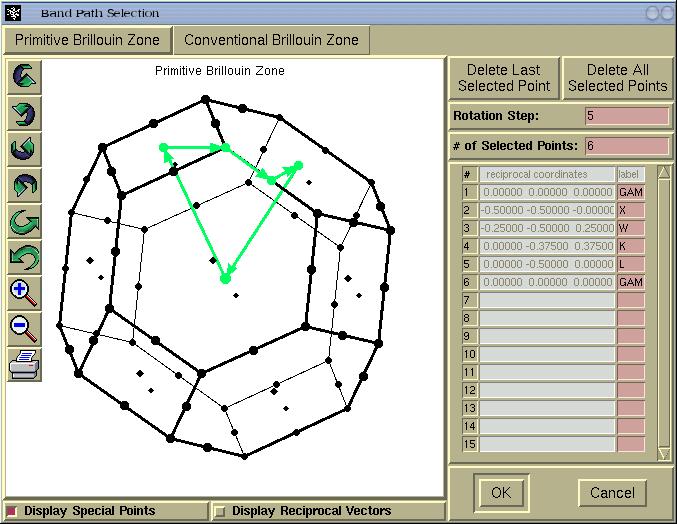
In above window we see two tabs entitled:
(i)
Primitive Brillouin Zone and (ii)
Conventional Brillouin zone. The latter is
provided only for informational purpose, namely, to see
the shape of the BZ tessellated according to the
conventional set of reciprocal vectors. Hence for real
applications we stick to
Primitive Brillouin
Zone and select a k-path by mouse-clicking a
special k-points. The BZ can be rotated by holding-down
left mouse button and dragging the mouse.
For a few Bravais lattice types, several common
k-points will be labelled automatically (thanks to
Peter Blaha), such as GAMMA, X, W, K, L points for
the fcc lattice. The automatic k-point labbeling
currently supports the following Bravais lattice
types:
- primitive cubic
- fcc
- bcc
- primitive tetragonal
- body centered tetragonal
- primitive orthorhomobic
When we are done with the k-points selection the
[OK] button should be pressed and a new window
will appear.
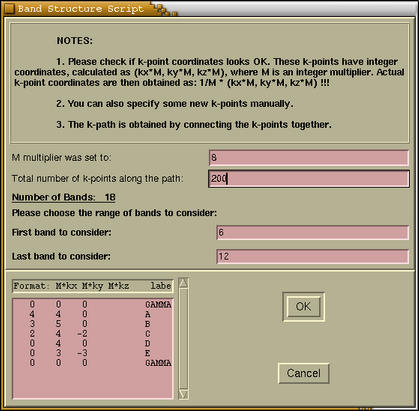
Here we specify the BAND parameters. Also
we can enter some additional k-points manually. After
pressing the
[OK] button the band structure is
calculated and displayed.
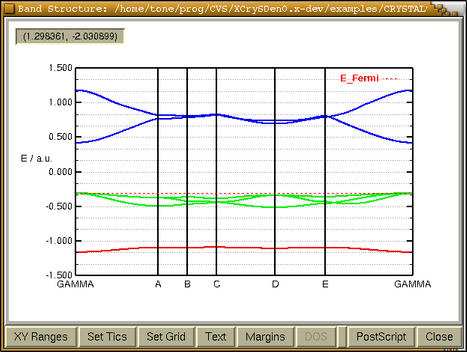
Some properties like charge density and
electrostatic potential can be represented in 2D or 3D
fashion, as contour plots or isosurfaces.
XCrySDen provides both
kinds of plotting. Moreover for periodic structures
this plots have periodic attribute, meaning that the
plots can be replicated in periodic directions.
BEWARE: it is suggested that only one 2D or 3D
calculation is performed per
XCrySDen run. For
example, rendering first the charge density, and later
on the electrostatic potentail might result in
Segmentation Fault. Instead, run the
XCrySDen twice. In first
run render the density, and in the second run the
potential.
In order to produce an isosurface of some property
(charge density, electrostatic potential) proceed via
Properties-->Isosurfaces
... menu. There one can choose among:
Charge
Density ,
Electrostatic Potential and
Difference
Maps .
When we enter
Charge
Density or
Electrostatic Potential option
under the
Properties-->Isosurfaces
... menu, then we first specify the grid
parameters in the
3D Map
- Grid Specification window. In this window we
find several entries. Their meaning is the following:
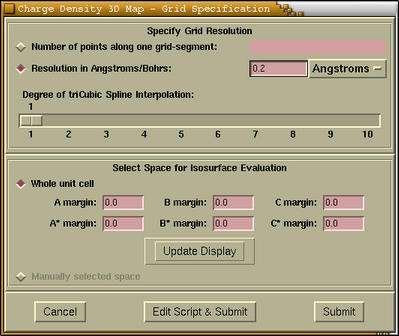
|
[Grid Resolution]
|
One can specify the resolution of the grid in
two ways: (i) by specifying the number of
points along one grid side ( n). Then
the number of all points in the grid is on
the order of n3. At least
for me this is not very intuitive. (ii) by
specifying the resolution in ANGSTROMS or
BOHRS. On the basis of my experiences the
quality of rendered isosurface can be
classified according to the resolution
as:
|
Grid Resolution / ANGSTROMS
|
Display of Isosurface
|
|
>0.3
|
poor
|
|
0.3-0.15
|
medium
|
|
<0.15
|
good
|
|
|
[Interpolation]
|
There is a possibility of interpolating a
grid by tri-cubic spline interpolation. At
this stage I would suggest to specify no
interpolation (degree=1), as it will be
possible to do that later.
|
|
[Space Selection]
|
By default the space comprised by the whole
unit cell is selected for the CRYSTALS. For
MOLECULES the space of the bounding-box is
selected instead. This box is just big enough
to hold the molecule. For SLABS and POLYMERS
it is a "linear combination" of both, namely,
unit cell in the periodic directions and
bounding-box in the non-periodic directions.
Then we
have the possibility to specify the margins.
The margins are in fractional units (either
with respect to unit cell or bounding-box -
depending on the periodicity of the
structure). Here is a simplified 2D example
that explains the meaning of this margins:
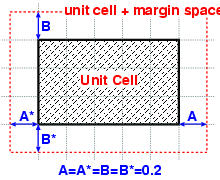
Important: Only after we
press the [Update button] the
currently selected space will be rendered as
transparent box. On the figure below we see
the unit-cell space selection with the
A=B=C=A*=B*=C*=0.2 margins.
|
There are three buttons on the bottom of
3D Map - Grid
Specification window. The function of these
buttons is the following:
|
[Cancel]
|
Cancels the process and closes the window
|
|
[Edit Script& Submit]
|
User will be able to modify the input script
manually in an editor, just before it is
submitted to calculation with CRYSTAL
properties module. Via this options
it is possible to alter the basis set,
density matrix or some other tuning. For
example: ...insert example....
Editor is chosen on the basis of EDITOR
environmental variable. If this variable does
not exists, the vi editor is
launched.
|
|
[Submit]
|
Submits the calculation to the CRYSTAL
properties module.
|
After CRYSTAL finishes calculating the 3D grid
of points of specified property the
Isosurface/Property-plane control window
appears, where we can control various parameters of
isosurface/property-plane rendering. The description of
this window and its parameters can be found
here.
When we enter the
Difference Maps under the
Properties-->Isosurfaces
... menu, then the following window
pops-up:
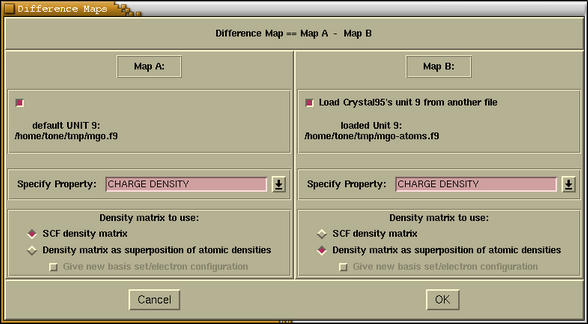
This window is divided in two parts:
Map A and
Map B. Later on, the
difference plots will be obtained by subtracting the
"Map B" from "Map A". For each map we should specify a
few parameters, such as: property to be calculated
(charge density, electrostatic potential) and some
settings about the density matrix. There is a
possibility to load the unit 9 from another file for
the
Map B (the
[Load Crystal95's unit 9
from another file] checkbutton). For example if we
want to render the charge deformation density (i.e.
charge density difference between an SCF density and an
atomic superposition densities) then in both maps we
select CHARGE DENSITY property and an
[SCF density
matrix] radiobutton for
Map A and
[Density matrix as superposition of atomic
densities] radiobutton for
Map B. Here it
is not possible to specify a new basis set for the
atomic superposition density, but this will be possible
later on, where we will be able to edit the CRYSTAL
input script just before it is submitted to
calculation. After pressing the information the
3D Map - Grid
Specification window appear. Further
information can be read
here.
In order to produce a contour or color-plane plot
of some property (charge density, electrostatic
potential) proceed via
Properties-->Properties on Plane
... cascade menu. There one can choose
among:
Charge
Density ,
Electrostatic Potential and
Difference
Maps .
When we enter
Charge
Density or
Electrostatic Potential option
under the
Properties-->Properties on Planes
... menu, then we first specify the grid
parameters in the
2D Map
- Grid Specification window that is shown
below. The process is analogues to the
3D grid specification, with the exception
that here we select a 2D region of space. The hints for
specifying the grid resolution can be found
here.
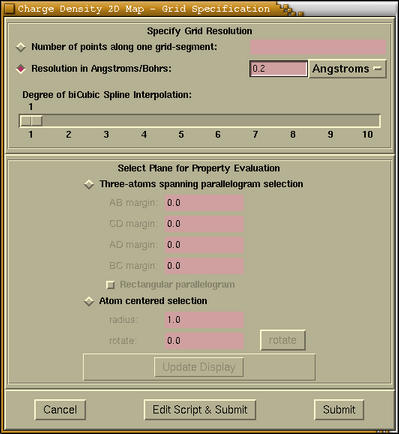
The second part (label
Select Plane
for Property Evaluation) of the
2D Map - Grid
Specification window is devoted to the selection
of 2D region of space. We can do that via two different
procedures, which are chosen by pressing the
corresponding radiobutton, that is either
[Three-atoms spanning parallelogram selection]
or
[Atom centered selection].
-
[Three-atoms spanning parallelogram
selection]
-
After clicking this radiobutton a selection window pops-up.
Here we select three atoms that lie on the desired
plane. Then we press the [Selected] button
on the selection window. After that we
press the [Update display] button on the
2D Map - Grid Specification window the
selected 2D region gets rendered as transparent
parallelogram. We can also specify the margins AB,
CD, AD, and BC. This margins are analogous to
margins A, A*, B, B*, C, C*
margins for the selection of 3D region. The updated
margins will be rendered only if we press the
[Update display] button. We have also a
possibility to force the selected parallelogram to
be rectangular. This is achieved by pressing
[Rectangular parallelogram] checkbutton.
-
[Atom centered selection]
-
The Atom centered selection is a two-step
procedure, where we should select (1) an atom
center and (2) a plane direction. After clicking
this radiobutton the following window appears:
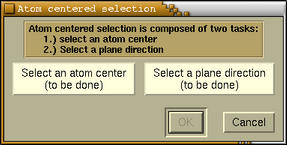
On this window two buttons named
[Select an atom center] and [Select a
plane direction] stand for the two tasks that
should be done. At the bottom of these buttons is
either a "(to be done)" or "(done)" label. These
remind us on the state of each task, namely, is the
task already done or yet to be done. The selection window will pop-up when
either one of these buttons is pressed. When
pressing the [Select an atom center] button
we should select one atom and press the
[Selected] button on selection window. On the other
hand when pressing the [Select a plane
direction] we should select three atoms that
define desired plane and press the
[Selected] button on the selection window. When both tasks
are completed we should press the [OK]
button on the Atom centered selection
window. Then we can press the [Update
Display] button on the selection window to render the
selected 2D region.
Three button are located at the bottom of
2D Map - Grid Specification
window. The description of their function can be found
here.
The process of creating
2D Difference
Maps is analogous to the creation of
3D Difference Maps. First we specify
the
difference map
parameters and then we select the
2D region of space, where the 2D
difference map will be calculated.








![[Figure]](img/xcrysden-picture-small-new.jpg)