Understanding, controlling, and designing the mechanical
properties of polymeric materials at the nanometer scale
has been a longstanding goal of many materials scientists and
engineers which has, for the most part, proved elusive.
New high-resolution force spectroscopy (HRFS) instruments
such as the surface force apparatus (SFA)[1], the atomic force microscope (AFM)[2],
the magnetic levitation force microscope (MLFM)[3], the molecular force probe (MFP)[4],
the biomembrane surface probe (BSP)[5], optical tweezers (OT)[6], the photonic
force microscope (PFM)[7], and nanoindentors (NI)[8], have made it routinely
possible to measure the physical properties of materials at the molecular
level and led to the new and expanding field of nanomechanics[9].
One of the most remarkable achievements in this area has been the
direct measurement of the elasticity of individual macromolecules[10-18],
In this project, we use some of the latest discoveries
in the field of single molecule force spectroscopy as models to
design biomimetic polymeric
systems with highly specific and controlled mechanical properties
at the molecular level. Although studies in this
area are important to many areas of microscopic
and macroscopic polymer science, we are primarily
motivated by recent developments and trends in the
field of nanotechnology. Functional nanometer-scale devices
have now become a reality; e.g. carbon nanotube "nanotweezers," [19]
molecular junctions consisting of an individual organic conducting
molecule between two gold leads[20], and metallic wires consisting
of a single row of gold atoms[21]. It is clear that miniaturized
devices will soon incorporate individual polymer chains to function
as molecular springs, switches, sensors, and motors, as well as serve
as the basic components for dynamic assemblies-just as they do in
biological systems. New design principles will be needed and a
comprehensive knowledge of the molecular-level forces and deformations
involved necessary for maximum efficiency and reliability. This
research will be the first step in producing working
synthetic macromolecular-based nanomechanical devices.
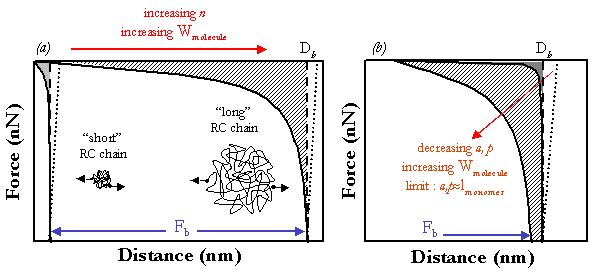
Molecular Elasticity of Random Coils.
As shown inthe Figure below, a single, random coil
(RC) polymer chain, such as polystyrene exhibits
a well-known purely entropic, reversible force (F)
versus separation distance or extension (D) behavior,
i.e. an initially linear, Gaussian regime, followed
by an increasingly nonlinear force which rises
asymptotically to the chain contour length, Lcontour.
Two of the most important parameters to consider are the
breaking strength, Fb, which is the force needed to break the weakest bonding
linkage in series along the polymer chain and the molecular toughness,
Wmolecule, which is the entropic deformational energy of the
polymer chain or area under the force versus distance curve :
Wmolecule=integral of F(D)dD integrated from 0 to the breaking distance, Db.
Fb is dependent on the pulling rate, rp, due to the fundamental kinetics
associated with force-driven bond dissociation pathways; i.e. as rp increases,
Fb increases[22]. Wmolecule is determined primarily by the amount of
intermediate- to high-stretch, nongaussian deformation that
takes place before break. Clearly, as Fb increases the chain can
be pulled farther and Wmolecule will also increase. Typically, a
polymeric linkage with both high strength, Fb, and high molecular
toughness, Wmolecule, is desirable and in this proposal, we will
concentrate on the latter parameter. There are two ways to increase
the molecular toughness of an individual random coil polymer chain;
by increasing the number of segments, n (or Lcontour)
or by decreasing the statistical segment length, a, or the
persistance length, p, (e.g. by placing in a bad solvent)
which increases the entropic resistance and shifts the curves
to higher forces.
However, changing n and a or p is not very effective in
increasing Wmolecule and in addition, provides little
control over the shape of the force versus distance curve.
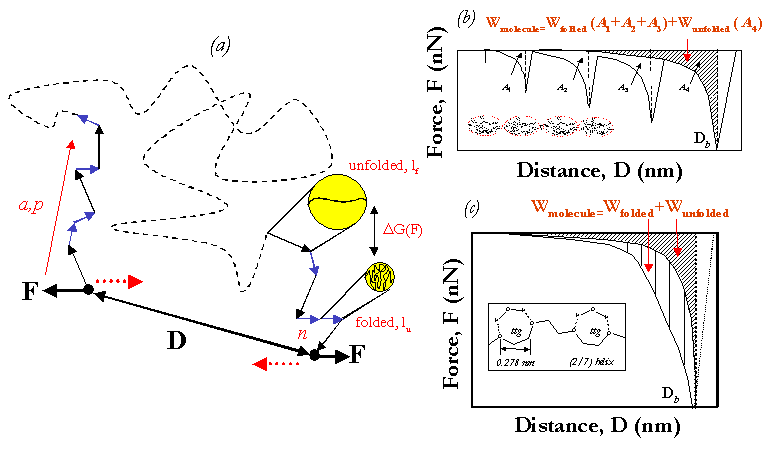
Molecular Elasticity of Organized Macromolecules.
It has been known for some time that nature ingeniously
controls the molecular elasticity of biological macromolecules
through the manipulation of local supramolecular structure
formed by noncovalent, secondary intramolecular interactions
(e.g. hydrogen bonding, van der Waals forces, hydrophobic interactions, etc.).
Recently, it was discovered that a general design principle exists in
which such macromolecules possess a local "modular" intrachain structure
composed of linear arrays of folded or looped domains[12].
When a modular polymer chain is extended,
initially it undergoes the standard entropic
elasticity described above which is controlled by n and a or p.
However, at intermediate to high extensions,
chemical-specific, strain-induced conformational
transitions of the subunits occur from the shorter,
folded state to an extended, unfolded state.
The result is almost always an increase in the molecular elasticity :
Wmolecule=Wfolded+Wunfolded.
where : Wunfolded is the purely entropic component of Wmolecule
in the absence of folding and Wfolded is the increase in
Wmolecule due to the strain-induced conformational transitions.
The increase in Wmolecule and shape of the force versus distance curve
now depend on many other parameters; most importantly the length of the
folded subunits, lf, relative to the length of the unfolded subunits,
lu, and the free energy difference between folded and unfolded subunits, dG [12].
The Figure above shows a schematic for the molecular elasticity
profile for the giant muscle protein, Titin, which has a filamentous,
modular structure consisting of a linear array of tightly folded
immunoglobin(Ig)- and fibronectin(FNIII)- like domains where lf is much less than lu[15].
Here we see that the force versus distance curve is characterized by a
unique "sawtooth" profile where each attractive peak corresponds to the
entropic uncoiling of an individual unfolded (mechanically denatured) domain.
The total molecular elasticity is then a summation of the areas under each
entropic uncoiling peak : Wmolecule=Wfolded(A1+A2+A3)+Wunfolded(A4).
Other proteins with similar modular structures such as Tenascin[16], Lustrin A[17],
and Spectrin[18] also exhibit sawtooth force profiles.
If lf is not much smaller than lu, e.g. double-stranded DNA[10], polysaacharides[11],
poly(ethylene oxide) (PEO)[12], poly(methacrylic acid) (PMAA)[13], and poly(vinyl alcolhol)
(PVA)[14], the individual unfolding events are not resolvable by
HRFS and instead one obtains an apparently smooth molecular
elasticity profile shifted to higher forces as shown in Figure above.
General Objectives and Design Strategies.
From these pioneering studies it has become clear that
a fundamental understanding and subsequent manipulation
of noncovalent intramolecular interactions will enable the
design of synthetic macromolecular systems with highly specific
and controlled mechanical properties at the molecular level.
Inspired by the biological concepts described above, here we will
use two general molecular design strategies for
improving the molecular toughness of synthetic macromolecules.
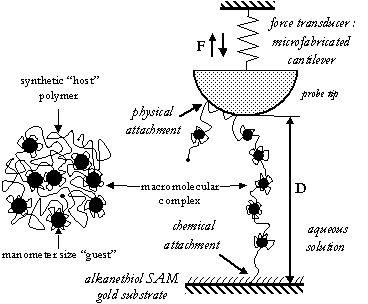
(I) The first involves the nonspecific, noncovalent complexation of a
synthetic "host" macromolecule with smaller, nanometer-size,
shape-persistant "guests" and (II) the second involves using more traditional
synthetic polymer chemistry routes.
The immediate goal is to create a high-toughness intersurface
macromolecular tether with an initially rapidly rising force
(high stiffness) followed by a relatively high-force "yield" which
takes place over large displacements where multiple, sacraficial,
noncovalent bonds are broken. In addition, we hope to be able to
design any type of molecular elasticity and toughness needed for a
particular application. For example, with the host-guest approach a
number of different parameters may be varied,
e.g. the size of guest relative to the size of host polymer chain,
Rguest/Rg (where Rg is the radius of gyration of the polymer chain),
the host backbone elasticity, chain stiffness, and supramolecular structure,
the strength and type of host-guest interaction which will control lu/lf,
the mechanical properties of guest (e.g. deformability, intramolecular
interactions, etc.), and the length of host chain segment between two
neighboring guests. In our experiments, chemical attachment of one
chain end of the host polymer to a substrate and sufficiently low
chain grafting densities will be achieved using a mixed monolayer
technique involving the co-chemisorption of mono(thiol, SH)-functionalized
polymers and a self-assembling alkanethiol monolayer (SAM) on gold [13].
The effectiveness of the proposed strategies will be evaluated in our
laboratory using high-resolution force spectroscopy. An individual
macromolecular complex will be tethered to the probe tip via
nonspecific, physisorption interactions and the extensional
nanomechanical properties (e.g. force, F, versus separation distance, D)
measured in aqueous solution.
REFERENCES:
1. Intermolecular and Surface Forces, 2nd. Ed., Israelachvili, J. N., Academic Press, 1992, pg. 169.
2. ōAtomic Force Microscope,ö Binnig, G.; Quate, C. F.; Gerber, Ch. Phys. Rev. Lett. 1986, 56 (9), 930-933.
3. (a) ōDesign For A 10 Picometer Magnetic Actuator,ö Gauthier-Manuel, B.; Garnier, L. Rev. Sci. Instrum. 1997, 68 (6), 2486-2489. (b) "Direct Measurement Of Short-Range Interaction Between A Tungsten Tip And A Mica Surface," Gauthier-Manuel, B. Europhys. Lett. 1992, 17, 195-200.
4. Asylum Research, Inc. (Santa Barbara, CA, USA) http://www.AsylumResearch.com/frame/index.html
5.ōSensitive Force Technique To Probe Molecular Adhesion And Structural Linkages At Biological Interfaces,ö Evans, E.; Ritchie, K.; Merkel, R. Biophys. J. 1995, 68, 2580-2587.
6. (a) ōObservation Of Radiation-Pressure Trapping Of Particles By Alternating Light-Beams,ö Ashkin, A.; Dziedzic, J. M. Phys. Rev. Lett. 1985, 54, 1245-1248. (b) ōObservation Of A Single-Beam Gradient Force Optical Trap For Dielectric Particles,ö Ashkin, A.; Dziedzic, J. M.; Bjorkholm, J. E.; Chu, S. Optics Letters 1986, 11, 288-290.
7. ōPhotonic Force Microscope Based On Optical Tweezers And Two-Photon Excitation For Biological Applications,ö Florin, E.-L.; Pralle, A.; Hoerber, J. K. H.; Stelzer, E. H. K. J. Struct. Bio. 1997, 119 (2), 202-211.
8. Nanoinstruments, Inc. (Oak Ridge, TN, USA), http://www.nanoinst.com/
9. see for example : Scanning Tunneling Microscopy and Spectroscopy: Theory, Techniques, and Applications, Bonnel, D. A. Ed.: VCH Publishers, New York, 1993, Burnham, N. A; Colton, R. J. pgs 191-249.
10. (a) ōStretch Genes,ö Austin, R. H.; Brody, J. P.; Cox, E. C.; Duke, T.; Volkmuth, W. Physics Today 1997, 50, 32-38. (b) ōDirect Mechanical Measurements Of The Elasticity Of Single Dna-Molecules By Using Magnetic Beads,ö Smith, S. B.; Finzi, L.; Bustamante, C. Science 1992, 258, 1122-1126. (c) ōOverstretching B-Dna: The Elastic Response Of Individual Double-Stranded And Single-Stranded Dna Molecules,ö Smith, S. B.; Cui, Y.; Bustamante, C. Science 1996, 271, 795-799. (d) ōEntropic Elasticity Of Lambda-Phage Dnaö Bustamante, C.; Marko, J. F.; Siggia, E. D.; Smith, S. B. Science 1994, 265, 1599-1600. (e) ōIonic Effects On The Elasticity Of Single Dna Molecules,ö Baumann, C. G.; Smith, S. B.; Bloomfield, V. A.; Bustamante, C. PNAS 1997, 94, 6185-6190. (f) ōDna: An Extensible Molecule,ö Cluzel, P.; LeBrun, A.; Heller, C.; Lavery, R.; Viovy, J.-L.; Chatenay, D.; Caron, F. Science 1996, 271, 792-794. (g) ōSequence-Dependent Mechanics Of Single Dna Molecules,ö Rief, M.; Clausen-Schaumann, H.; Gaub, H. E. Nature Struct. Bio. 1999, 6 (4), 346.
11. (a) ōSingle Molecule Force Spectroscopy On Polysaccharides By Atomic Force Microscopy,ö Rief, M.; Oesterhelt, F.;Heymann, B.; Gaub, H. E. Science 1997, 275, 1295-1297. (b) ōSingle-Molecule Force Spectroscopy On Xanthan By Afm,ö Li, H.; Rief, M.; Oesterhelt, F.; Gaub, H. E. Adv. Mater. 1998, 4, 316-319. (c) ōForce Spectroscopy On Single Xanthan Molecules,ö Li, H.; Rief, M.; Oesterhelt, F.; Gaub, H. E. Appl. Phys. A 1999, 68, 407-410. (d) ōSingle-Molecule Force Spectroscopy On Polysaccharides By Afm - Nanomechanical Fingerprint Of Alpha-(1,4)-Linked Polysaccharides,ö Li, H.; Rief, M.; Oesterhelt, F.; Gaub, H. E.; Zhang, X.; Shen, Chem. Phys. Lett. 1999, 305, 197-201. (e) ōAtomic Levers Control Pyranose Ring Conformations,ö Marszalek, P. E.; Pang, Y.-P.; Li, H.; Yazal, J. E.; Oberhauser, A. F.; Fernandez, J. M. PNAS (Biophys.) 1999, 96, 7894.
12. ōSingle Molecule Force Spectroscopy By Afm Indicates Helical Structure Of Poly(Ethylene-Glycol) In Water,ö Oesterhelt, F.; Rief, M.; Gaub, H. E. New J. Physics 1999, 1, 6.1-6.11.
13. ōEntropic Elasticity Of Single Polymer Chains Of Poly(Methacrylic Acid) Measured By Atomic Force Microscopy,ö Ortiz, C.; Hadziioannou, G. Macromolecules 1999, 32, 780-787.
14. ōHydrogen Bonding Governs The Elastic Properties Of Poly(Vinyl Alcohol) In Water: Single-Molecule Force Spectroscopic Studies Of Pva By Afm,ö Li, H.; Zhang, W.; Xu, W.; Zhang, X. Macromolecules 2000, 33(2), 465-469.
15. (a) ōReversible Unfolding Of Individual Titin Immunoglobulin Domains By Afm,ö Rief, M.; Gautel, M.; Oesterhelt, F.; Fernandez, J. M.; Gaub, H. E. Science 1997, 276, 1109-1112. (b) ōFolding-Unfolding Transitions In Single Titin Molecules Characterized With Laser Tweezers,ö Kellermayer, M. S. Z.; Smith, S. B.; Granzier, H. L., Bustamente, C. Science 1997, 276, 1112-1116. (c) ōElastic Properties Of Single Titin Molecules Made Visible Through Fluorescent F-Actin Binding,ö Kellermayer, M. S. Z.; Granzier, H. L. Biochem. Biophys. Res. Commun. 1996, 221, 491-497. (d) ōElasticity And Unfolding Of Single Molecules Of The Giant Muscle Protein Titin,ö Tskhovrebova, L.; Trinick, J.; Sleep, J. A.; Simmons, R. M. Nature 1997, 387, 308-312. (e) ōProtein Biophysics - Stretching Single Protein Molecules: Titin Is A Weird Spring,ö Erickson, H. P. Science 1997, 276, 1090-1092. (f) ōMechanical Unfolding Intermediates In Titin Modules,ö Marszalek, P. E.; Lu, H.; Li, H.; Carrion-Vazquez, M.; Oberhauser, A. F., Schulten, K.; Fernandez, J. M. Nature 1999, 402 (4), 100. (g) ōAtomic Force Microscopy Captures Length Phenotypes In Single Proteins,ö Carrion-Vazquez, M.; Marszalek, P. E.; Oberhauser, A. F., Fernandez, J. M. PNAS (Biophys.) 1999, 96, 11288. (h) ōAtomic Force Microscopy Reveals The Mechanical Design Of A Modular Protein,ö Li, H.; Oberhauser, A. F., Fowler, S. B.; Clarke, J.; Fernandez, J. M. PNAS (Biophys.) 2000, 97, 6527-6531. (i) ōMechanical And Chemical Unfolding Of A Single Protein: A Comparison,ö Carrion-Vazquez, M.; Oberhauser, A. F.; Fowler, S. B.; Marszalek, P. E.; Broedels, S. E.; Clarke, J.; Fernandez, J. M. PNAS (Biophys.) 1999, 96, 3694.
16. ōThe Molecular Elasticity Of The Extracellular Matrix Protein Tenascin,ö Oberhauser, A. F.; Marszalek, P. E.; Erickson, H. P.; Fernandez, J. M. Nature 1998, 393, 181.
17. ōMolecular Mechanistic Origin Of The Toughness Of Natural Adhesives, Fibres And Composites,ö Smith, B. L.; Schaffer, T. E., Viani, M., Thompson, J. B., Frederick, N. B., Kindt, J., Belcher, A., Stucky, G. D., Morse, D. E., Hansma, P. K. Nature 1999, 399, 761-763.
18. ōSingle Molecule Force Spectroscopy Of Spectrin Repeats: Low Unfolding Forces In Helix Bundles,ö Rief, M.; Pascual, J.; Saraste, M.; Gaub, H. E. J. Mol. Biol. 1999, 286, 553-561.
19. ōNanotube Nanotweezers,ö Kim, P.; Lieber, C. M. Science 1999, 286, 2148.
20. ōConductance Of A Molecular Junction,ö Reed, M. A.; Zhou, C.; Muller, C. J.; Burgin, T. P.; Tour, J. M. Science 1997, 278, 252.
21. ōFormation And Manipulation Of A Metallic Wire Of Single Gold Atoms,ö Yanson, A. I.; Bollinger, G. R.; van den Brom, H. E.; Agrait, N.; van Ruitenbeek, J. M. Nature 1998, 395, 783.
22. (a) "Probing Nanoscale Adhesion And Structure At Soft Interfaces," Ritchie, K. Ph.D. Thesis, University of British Columbia, 1998. (b) ōEnergy Landscapes Of Receptor-Ligand Bonds Explored With Dynamic Force Spectroscopy,ö Merkel, R.; Nassoy, P.; Leung, A.; Ritchie, K.; Evans, E. Nature 1999, 397, 50. (c) ōLooking Inside Molecular Bonds At Biological Interfaces With Dynamic Force Spectroscopy,ö Evans, E. Biophys. Chem. 1999, 82, 83-97. (d) ōEnergy Landscapes Of Biomolecular Adhesion And Receptor Anchoring At Interfaces Explored With Dynamic Force Spectroscopy,ö Evans, E. Faraday. Disc. 1998, 111, 1. (e) ōStrength Of A Weak Bond Connecting Flexible Polymer Chains,ö Evans, E.; Ritchie, K. Biophys. J. 1999, 76 (5), 2439.
23. (a) "Mechanical Properties Of Mother Of Pearl In Tension," Currey, J. D. Proc. R. Soc. Lond. B 1977, 196, 443-463. (b) ōThe Mechanical Design Of Nacre,ö Jackson, A. P.; Vincent, J. F.; Turner, R. M.; Proc. R. Soc. Lond. B 1988, 234, 415-440.