The
genome of a species encodes genes and other functional elements, interspersed
with non-functional nucleotides in a single uninterrupted string of DNA. Recognizing protein-coding genes relies on
finding stretches of nucleotides free of stop codons (called Open Reading
Frames, or ORFs) that are too long to have likely occurred by chance. Since stop codons occur at a
frequency of roughly 1 in 20 in random sequence, ORFs of at least 60 amino
acids will occur frequently by chance (5% under a simple Poisson model) and
even ORFs of 150 amino acids will appear by chance in a large genome (0.05%).
This poses a huge challenge for higher eukaryotes in which genes are typically
broken into many, small exons (on average 125 nucleotides long for internal
exons in mammals27).
The basic
problem is distinguishing real genes – those
ORFs encoding a translated protein product – from spurious ORFs – the remaining ORFs whose presence is simply due to
chance. The current public catalogue of
yeast genes lists 6062 predicted ORFs that could theoretically encode proteins
of at least 100 amino acids. Only
two-thirds of these have been experimentally validated (known), and the remaining ~2000 ORFs are currently annotated as hypothetical. The total number of real protein-coding genes has been a subject
of considerable debate, with estimates ranging from 4,800 to 6,400 genes (in
mammalian genomes, estimates have ranged from 28,000 to more than 120,000
genes).
In
this chapter, we use
the comparative information to recognize real genes based on their conservation
across evolutionary time. With the
availability of genome-wide alignments across the four species, we first
examined the different ways by which sequences change in known genes and in
intergenic regions. The alignments of
known genes revealed a clear pressure to preserve protein-coding
potential. We constructed a computational
test for reading frame conservation (RFC) and used it to revisit the annotation
of yeast. We showed that more than 500
previously annotated ORFs are not meaningful and discovered 43 novel ORFs that
were previously overlooked. We
additionally refined the gene structure of hundreds of genes, including
translation start, stop, and exon boundaries.
We show that our method has high sensitivity and specificity, and
suggest changes that affect nearly 15% of yeast genes.

We examined the different types of conservation in genes and intergenic
regions. We used the 1-to-1 orthologous
anchors (see Chapter 1) to construct a nucleotide-level alignment of the
genomes. The strong conservation of local
gene order and spacing (Figure 2.1) allowed us to construct genome-wide
multiple alignments. We aligned each
gene together with its flanking intergenic regions using CLUSTALW38 for the multiple alignments
across the four species. When
sequence gaps were present in one or more species, we constructed the alignment
in multiple steps. We first aligned the
gapless species creating a base alignment.
Then we aligned each portion of a partially covered ortholog onto the
base alignment, and constructed a consensus for each species based on the
individually aligned portions. We
marked missing sequence between contigs by a dot and disagreeing overlapping
contigs by N. Finally, we constructed a
multiple alignment of the four species by merging the piece-wise alignments. With sequence alignments at millions of
positions across the four species, it is possible to obtain a precise estimate
of the rate of evolutionary change, including substitutions and
insertion-deletions (indels), in the tree connecting the species. We counted transitions, transversions,
insertions and deletions within these alignments and used these to estimate the
rate of evolutionary change between the species. We counted the rate of
synonymous and non-synonymous substitutions for every protein coding gene to
find evidence of positive selection.
The detailed results will be described in chapter 6.
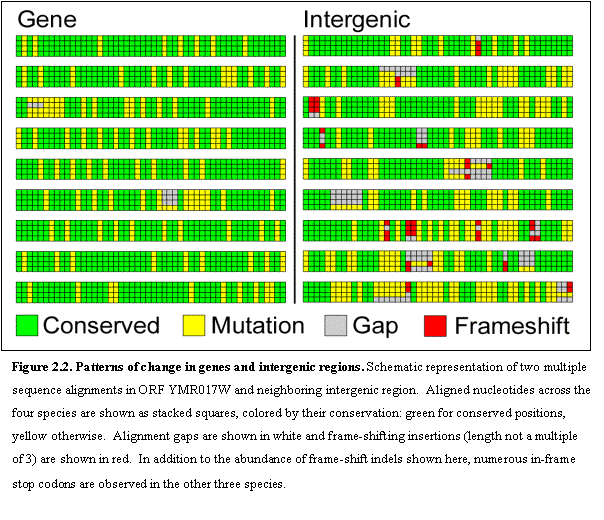
We compared the rate of sequence change at aligned sites across the four
species in intergenic and genic (protein-coding) regions (Figure 2.2). We found radically different types of
conservation. Intergenic regions
typically showed short stretches between 8 and 10 bases of near-perfect
conservation, surrounded by non-conserved bases, rich in isolated gaps. Protein-coding genes on the other hand were
much more uniform in their conservation, and typically differed in the
largely-degenerate third-codon position.
The proportion of sites corresponding to a different nucleotide in at
least one of the three species is 58% in intergenic regions but only 30% in genic
regions – a difference of ~2-fold. The
difference becomes much greater when one considers the gapped positions in
alignments, representing insertion and deletion events (indels). The proportion of indels is 14% in
intergenic regions, but only 1.3% in genic regions. The contrast is even sharper for indels whose length is not a
multiple of three. These would disrupt
the reading frame of a functional protein-coding gene, and are detrimental when
they occur in real genes, unless they are compensated by a nearby indel that
restores the reading frame.
Frame-shifting gaps are found in 10.2% of aligned positions in
intergenic regions, but only in 0.14% of positions in genic regions, a 75-fold
strong separation. We used these alignment properties to recognize
real genes.
We developed a Reading Frame Conservation (RFC) test to
classify each ORF in S. cerevisiae as
biologically meaningful or not, based on the proportion of the ORF over which
reading frame is locally conserved in each of the other three species. Each species with an orthologous alignment
cast a vote for accepting or rejecting the ORF, and the votes were tallied to
reach a decision for that ORF.

We
evaluated the percent of nucleotides that are in the same frame within
overlapping windows of the alignment.
For every such window, we labeled each nucleotide of the first sequence
by its position within a codon, as 1, 2 or 3 in order, starting at codon offset
1. We similarly labeled the nucleotides
of the second sequence, but once for every start offset (1, 2, or 3). We then
counted the percentage of gapless positions in the alignment that contained the
same label in both aligned species, and selected the maximum percentage found
in each of the three offsets of the second sequence (Figure 2.3). The final RFC value for the ORF was
calculated by averaging the percentages obtained at overlapping windows of 100
nucleotides starting every 50 nucleotides.
For overlapping ORFs in the S.
cerevisiae genome (n = 948), the
RFC was calculated only for the portion unique to each overlapping ORF. For spliced genes (n = 240), the RFC was calculated only on the largest exon.
We found that the distribution of frame conservation within
each species is bimodal, and we chose a simple cutoff for each species, 80% for
S.paradoxus, 75% for S.mikatae and 70% for S.bayanus. If the RFC of the best hit was above the cutoff, a species voted
for keeping the ORF tested. If the RFC
was below the cutoff and the hit was trusted as orthologous, the species voted
for rejecting the tested ORF. Finally,
if no orthologous hit could be found due to coverage, a species abstained from
voting. We calculated a score between
–3 and +3 for every ORF based on the number of species that accepted it (+1)
and the number of species that rejected it (-1). We kept all ORFs with a score of 1 or greater, and rejected all
ORFs with a score of –1 or smaller. We
manually inspected the remaining ORFs.

We also applied this test to 3966 annotated ORFs with associated gene names
(Table 2.4). These have been studied
and named in at least one peer-reviewed publication, and are likely to be
represent real genes. Only 15 of these
(0.38%) were rejected (KRE20, KRE21,
KRE23, KRE24, VPS61, VPS65, VPS69, BUD19, FYV1, FYV2, FYV12, API2, AUA1, ICS3,
UTR5, YIM2). We inspected these
manually and concluded that all were indeed likely to be spurious. Most lack
experimental evidence. For the remainder, reported phenotypes associated with
deletion of the ORF seems likely to be explained by fact that the ORF overlaps
the promoters of other known genes.
To investigate the power of the approach to reject spurious
ORFs, we also applied it to a set of controls sequences consisting of 340
intergenic sequences in S. cerevisiae
with lengths similar to the ORFs tested (Table 2.4). About 96% were rejected as
having conservation properties incompatible with a biologically meaningful ORF,
showing that the test has high sensitivity. Of the remaining 4% that were not
rejected, close inspection shows that three-quarters appear to contain true
ORFs. Some define short ORFs with conserved start and stop codons in all four
species and others extend S. cerevisiae
ORFs in the 5’- or 3’-direction in each of the other three species. Thus, at
most 1% of true intergenic regions failed to be rejected by the RFC test.
The conservation-based gene identification algorithm we
proposed has thus high sensitivity and specificity. In the next section, we apply it systematically for de-novo gene
identification in S. cerevisiae.
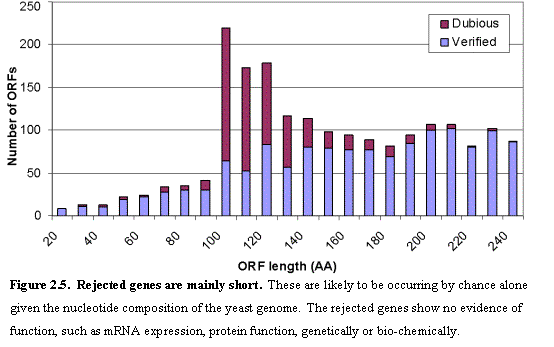
When the yeast genome sequence was completed23, 6275 ORFs were identified in
the nuclear genome that could theoretically encode proteins encoding at least
100 amino acids and that do not overlap a longer ORF by more than half of their
length (Figure 2.5). SGD has since updated the catalog based on complete
resequencing and re-annotation of chromosome III, re-analysis of other
chromosomes and reports in the scientific literature. This resulted in a
current version (as of May 2002) with 6062 ORFs ≥ 100 amino acids,
consisting of 3966 ‘named’ genes (described in at least one publication) and 2096
‘uncharacterized’ ORFs. SGD also includes a small collection of ORFs < 100
amino acids (see below).
We sought to apply the RFC test to all 6062 ORFs in SGD. A
total of 117 could not be analyzed because they were almost completely
contained within an overlapping ORF (99 cases, with average non-overlapping
portion = 12 bp) or because an orthologous region could not be unambiguously
defined in any of the species (18 cases).
Of the 5945 ORFs tested, the analysis strongly validated 5550 ORFs. The
vote was unanimous in 5458 (~98%) of cases.
In the remaining cases, a valid gene appears to have degenerated in one
of the four species. A total of 367
ORFs were strongly rejected. These
rejections were unanimous in 63% of cases.
In most of the remaining cases, S.
paradoxus was too closely related to S.
cerevisiae to have accumulated enough frameshifts to allow definitive
rejection. The analysis deadlocked (one
confirmation, one rejection, one abstention) for 28 ORFs (0.5%). We inspected these, together with the 117
cases that could not be analyzed due to overlaps and found convincing evidence
(based on conservation of amino acids, start and stop codons, and presence of
indels), that 20 are valid protein coding genes and 105 are spurious. We were
unable to reach a judgment in the remaining 20 cases. Overall, a total of 5570 ORFs were accepted, 472 ORFs were
rejected, and 20 remain ambiguous.

The vast majority of the rejections (96%) involve uncharacterized ORFs (for an
example see Figure 2.6). SGD reports no
compelling biological evidence (such as changes in mRNA expression) to suggest
that these ORFs encode a true gene.
Most of these overlap another well-conserved ORF, but show many
insertions and deletions in the non-overlapping portion. The remainder tend to be small (median = 111
aa, with 93% ≤ 150 aa) and show atypical codon usage23,39,40. Figure 2.6 illustrates the
case of an ORF of 333 bp that is clearly biologically meaningless. The orthologous sequence in all four species
is laden with frameshifts (as well as stop codons). Only one rejected ORF, YBR184W, appears to represent a true gene
that fails the RFC test because it is evolving very rapidly (see section 6.6).
In summary, the Reading Frame Conservation (RFC) test
allowed a major revisiting of the yeast genome annotation. By observing the pattern of indels in the
multiple alignment of predicted ORFs, it allowed us to automatically classify
them as biologically meaningful or spurious.
It reached a decision automatically in 98% of cases, accepting 99% of
named ORFs and rejecting 99% of real intergenic regions, showing strong
sensitivity and specificity. It
resulted in a drastic reduction of the yeast gene count, rejecting nearly 500
ORFs. We next use the comparative
information to refine the boundaries of ORFs.
Comparative genome analysis not only improves the
recognition of true ORFs, it also yields much more accurate definitions of gene
structure – including translation start, translation stop and intron
boundaries. We used the comparative
data to identify sequencing errors and refine the boundaries of true genes. Previous annotation of S. cerevisiae has defined the start of translation as the first
in-frame ATG codon. However, the actual start of translation could lie 3’ to
this point, and the earlier in-frame ATG may be due to chance. Alternatively, if sequencing errors or
mutations have obscured an earlier in-frame ATG codon, the true translation
start could lie 5’ to this point. Similarly, the annotated stop codon could be
erroneously annotated, due to sequencing errors. Identifying the correct gene boundaries is important for many
reasons, both experimental (for example to construct gene probes), as well as
computational (for example to search for regulatory motifs).
We examined the multiple alignment of unambiguous ORFs to
identify discrepancies in the predicted start and stop codons across the four
species. We searched for the first
in-frame ATG in each species and compared it to the annotated ATG in S. cerevisiae. In the S. cerevisiae
start was not conserved, we automatically suggested a changed translation start
if a subsequent in-frame ATG was conserved in all species and was the first
in-frame ATG in at least one species.
Otherwise, we searched for a conserved ATG 5’ to that point. Similarly, we suggested changes in stop
codons when a common stop in all other species disagreed with the S. cerevisiae annotation. We manually inspected the alignments to
confirm that the suggested start and stop boundary changes agreed with
conservation boundaries. We identified
merges of consecutive S. cerevisiae
ORFs, when they unambiguously matched a single ORF in at least one other
species, and when their lengths added up to the length of the matching
ORF.
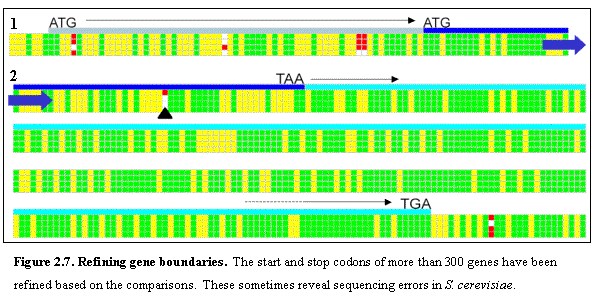
We identified 210 cases in which the presumed translational start in S. cerevisiae does not correspond to the
first in-frame start codon in at least two of the three other species (Figure
2.7 panel 1). In the vast majority of
these cases, inspection of the sequence alignments provides strong evidence for
an alternative conserved position for the translational start, either 3’ or 5’
to the previous annotation. We observed
a lower overall conservation as well as frame-shifting indels outside the new
boundaries. Similarly, we identified
330 cases in which the presumed translational stop codon in S. cerevisiae does not correspond to the
first in-frame stop codon in at least two of the three species. In ~25% of
these cases, the other three species share a common stop codon and a single
base change to the S. cerevisiae
sequence would result in a stop codon in the corresponding location (Figure 2.7
panel 2). The remaining 75% of cases
appear to represent true differences in the location of the translational stop
across the species. Thus, stop codons appear to show more evolutionary
variability in position than start codons.
We also developed methods for the automatic detection of
frame-shifting sequencing errors. When
regions of the multiple alignment shifted from one well-conserved reading frame
to another well-conserved reading frame, we pinpointed regions of potential
sequencing errors in each of the species.
A number of these were detected in the reference sequence of S. cerevisiae. We confirmed 32 of these computational predictions by
resequencing and found that in each case the published sequence was in error,
and that the predicted erroneous nucleotide was always within a few base pairs
from the experimentally confirmed sequencing error.
We identified 32 cases where two adjacent ORFs in S. cerevisiae are joined into a single
ORF in all three other species. In
every case, a single nucleotide change would suffice to join the ORFs in S. cerevisiae (either a substitution
altering a stop codon or an indel altering the reading frame). In principle, these cases could represent
errors in the genome sequence, mutations private to the sequenced strain S288C,
or substitutions fixed in S. cerevisiae.
We examined 19 cases by resequencing the relevant region in S288C. Our results
revealed an error in the published sequence in 11 cases (establishing that
there is a single ORF in S288C) and confirmed the published sequence in the
remaining 7 cases. Sequencing of
additional strains will be required to determine whether these remaining cases
represent differences in S288C alone or in S. cerevisiae in general.
We also found two named ORFs (FYV5 and CWH36) that pass
the RFC test and cause phenotypes when deleted, but show no significant protein
similarity across the four species. In both cases, inspection reveals that the
opposite strand encodes a protein that shows strong amino acid conservation.
(The latter gene has two introns, increasing the count of doubly spliced genes
to 8.) In each case, we postulate that the protein responsible for the reported
deletion phenotype is encoded on the opposite strand.
All merges and boundary refinements suggested specific changes
to the nucleotide sequence of S.
cerevisiae (except 3’ changes of translation start that required no
change). To validate our predictions,
we re-sequenced the sites of predicted sequence discrepancies. We used both forward and reverse reads in
two different PCR reactions spanning the site.
We examined 4 cases in which the comparative data suggested an earlier
start codon and found, by resequencing, that all correspond to errors in the
published sequence of S288C. We
examined 17 such cases and found that 15 are explained by errors in the
published sequence of S288C.
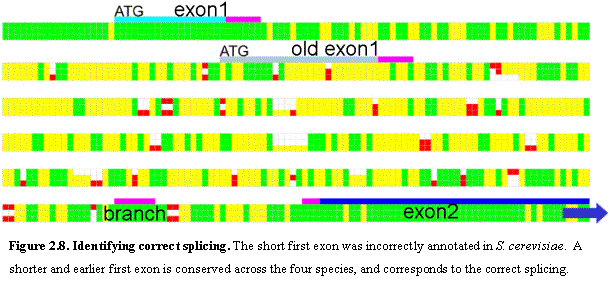
New Introns. We
also examined the conservation of introns in the yeast genome. We studied 218
of the 240 ORFs reported in SGD to contain at least one intron (omitting the
rest primarily due to lack of an orthologous alignment). In 92% of cases, the donor, branchpoint, and
acceptor sites were all strongly conserved with respect to both location and
sequence. Moreover, exon boundaries closely demarcated the domains of sequence
conservation as measured by both nucleotide identity and absence of
indels. Discrepancies were found in 17
cases, of which at least 9 strongly suggest that the previous annotation is
incorrect. Five identify a new first exon (Figure 2.8) and four predict that a
previously annotated intron is spurious.
We then sought to identify previously unrecognized introns by searching the
S. cerevisiae genome for conserved splicing signals. We searched for conserved and proximal
splice donor and branch signals and manually inspected the resulting
alignments. Having constructed multiple
alignments of ORFs and flanking intergenic regions, we searched for conserved
splicing signals. We used 10 variants
of splice donor signals (6-7bp) and 8 variants of branch site signals (7bp) that
are found in experimentally validated S.
cerevisiae introns41. We searched each species independently but required that
orthologous signals appear within 10 bp from each other in the multiple
alignment of the region. We also
required that branch and donor be no more than 600bp apart, which is the case
for 90% of known S. cerevisiae introns. We then inspected the multiple alignment
surrounding the conserved signals for three properties: (1) a conserved
acceptor signal, [CT]AG, 3’ of the branch site (2) high RFC 5’ of the donor
signal and 3’ of the acceptor signal.
(3) low RFC within the intron.
Roughly half of the conserved donor/branch pairs met our additional
requirements.
We predict 58 novel introns. Fifty cases affect the
structure of known genes (defining new 5’-exons in 42 cases, 3’-exons in 7
cases and an internal splice in one case) and two indicate the presence of new
genes. The relationship of the apparent splice signals to existing genes is
unclear for the remaining six cases. We visually inspected our predictions and
compared our results to experimental studies by Ares and colleagues that
identified new introns using techniques such as microarray hybridization41. Of our 58 predicted introns,
20 were independently discovered by this group. Of the four annotated introns
predicted to be spurious, all four show no experimental evidence of splicing.
Our remaining predictions are currently being tested in collaboration with Ares
and colleagues.
The power of our method was limited for small ORFs. Smaller regions may indeed show lack of
indels due to chance, and hence a high reading frame conservation score may not
be meaningful.
We tested 141 ORFs encoding 50-99 amino acids for which some
biological evidence has been published and are reported in SGD. Applying the
RFC test and inspecting the results, we conclude that 120 appear to be true
genes, 18 appear to be spurious ORFs and 3 remain unresolved. SGD also lists 32
ORFs encoding < 50 aa. We did not undertake a systematic search for all such
ORFs, because control experiments showed that the RFC test lacked sufficient
power to prove the validity of such small ORFs (see below). However, it is able to reject 7 of the 32
ORFs as likely to be spurious. Our
yeast gene catalogue thus contains 188 short genes (<100 aa), of which 43
are novel.
To evaluate the predictive power of the RFC test for small
ORFs, we additionally tested for presence of in-frame stop codons in the other
species. When a small ORF in S. cerevisiae showed a strong overall
frame conservation, we measured the length of the longest ORF in the same
orientation in each orthologous locus. We measured the percent of the S. cerevisiae length that was open in
each species (no stop codons), and took the minimum of the three percentages
(OPEN) across the three additional species.
When the reading frame was open in each of the other species, the
lengths found were identical to that of S.
cerevisiae, and OPEN was 100%. When
OPEN was below 80%, we concluded that stop codons appeared in the orthologous
sequence, and therefore that the RFC test falsely accepted a segment that did not
correspond to a true gene. We observed
the distribution of OPEN for different values of RFC. For S. cerevisiae ORFs
between 50 and 100 amino acids (aa), selecting for high RFC automatically
selected for high OPEN, and we estimated the test has high specificity. For ORFs between 30 and 50 aa however, only
a small portion of the ORFs with high RFC show a high OPEN, and we conclude
that the lack of indels within the small interval considered is not due to
selective pressure, but instead lack of evolutionary distance between the
species aligned.
We further systematically searched the remainder of the S. cerevisiae genome and evaluated all ORFs in this size range.
Control experiments demonstrated that the RFC test has high power to
discriminate reliably between valid and spurious ORFs in this size range. The
genome contains 3161 such ORFs, nearly all are readily rejected by the RFC
test. However, 43 novel genes were identified. These ORFs not only pass the RFC
test, but they also have orthologous start and stop codons. Five of these have
been reported in the literature subsequent to the SGD release studied here
Based on the analysis above, we propose a revised yeast gene
catalog consisting of 5538 ORFs ≥ 100 amino acids. This reflects the
proposed elimination of 503 ORFs (366 from the RFC test, 105 by manual
inspection and 32 through merger). A total of 20 ORFs in SGD remain unresolved.
Complete information about the gene catalog is provided in 29 and will be discussed more
fully in a subsequent manuscript in collaboration with SGD and other yeast
investigators. The revised gene count
is consistent with at least two recent predictions based on light shotgun
coverage of related species4,5. We believe that this represents a reasonably accurate description
of the yeast gene set, because the analysis examines all ORFs ≥ 100 amino
acids, the methodology has high sensitivity and specificity and the evidence is
unambiguous for the vast majority of ORFs. Nonetheless, some errors are likely
to remain. The results could be confirmed and remaining uncertainties resolved
by sequencing of additional related yeast species, as well as by other
experimental methods.
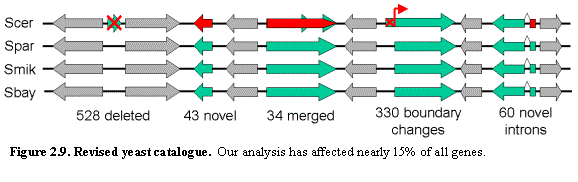
Despite the intensive study of S. cerevisiae to date, comparative genome analysis points to the
need for a major revision of the yeast gene catalog affecting more than 15% of
all ORFs (Figure 2.9). The results
suggest that comparative analysis of a modest collection of species can permit
accurate definition of genes and their structure. Comparative analysis can complement the primary sequence of a
species and provide general rules for gene discovery that do not rely solely on
known splicing signals for gene discovery.
Previous studies have shown that such methods are also applicable to the
understanding of mammalian genes42. The ability to observe the evolutionary pressures that nucleotide
sequences are subjected to radically changes our power for signal discovery.