| |
Sequin Quick Guide |
| Sequin | Entrez | BLAST | OMIM | Taxonomy | Structure |
Sequin for Database Submissions and Updates:
A Quick Guide
Jonathan Kans, Colombe Chappey, Jinghui Zhang, Tatiana Tatusov, and James
Ostell
National Center for Biotechnology Information, National Library of
Medicine, NIH
Sequin is a stand-alone software tool developed by the NCBI for submitting and updating sequences to the GenBank, EMBL, and DDBJ databases. Sequin has the capacity to handle long sequences and sets of sequences (segmented entries, as well as population, phylogenetic, and mutation studies). It also allows sequence editing and updating, and provides complex annotation capabilities. In addition, Sequin contains a number of built-in validation functions for enhanced quality assurance.
This overview is intended to provide a quick guide to Sequin's capabilities, including automatic annotation of coding regions, the graphical viewer, quality control features, and editing features. We suggest that you read this entire document before beginning your Sequin submission. More detailed instructions on these and other functions can be found in Sequin's on-screen Help file, also available on the World-Wide Web from the Sequin home page at:
http://www.ncbi.nlm.nih.gov/Sequin/
E-mail help is also available from info@ncbi.nlm.nih.gov
Basic Sequin Organization
Sequin is organized into a series of forms to (1) enter submitting authors, (2) enter organism and sequences, (3) enter information such as strain, gene, and protein names, (4) view the complete submission, and (5) edit and annotate the submission. The goal is to go quickly from raw sequence data to an assembled record that can be viewed, edited, and submitted to your database of choice.
Advance through the pages that comprise each form by clicking on labeled folder tabs or the Next Page button. After the basic information forms have been completed and the sequence data imported, Sequin provides a complete view of your submission, in your choice of text or graphic format. At this point, any of the information fields can be easily modified by double-clicking on any area of the record, and additional biological annotations can be entered by selecting from a menu.
Sequin has an on-screen Help file that is opened automatically when you start the program. Because it is context-sensitive, the Help text will change and follow your steps as you progress through the program. A "Find" function is also provided.
Preparing Nucleotide and Amino Acid Data
Sequin normally expects to read sequence files in FASTA format. Note that most sequence analysis software packages include FASTA or "raw" as one of the available output formats. Population, phylogenetic, and mutation studies can also be entered in PHYLIP, NEXUS, MACAW, or FASTA+GAP formats.
See http://www.ncbi.nlm.nih.gov/Sequin/faq.html#Orgnameforphyl or "Sample Data Files" for detailed examples of each of the various input data formats.
Prepare your sequence data files using a text editor, and save in ASCII text format (plain text). If your nucleotide sequence encodes one or more protein products, Sequin expects two files, one for the nucleotides and one for the proteins.
FASTA format is simply the raw sequence preceded by a definition line. The definition line begins with a > sign, and is followed immediately by a name for the sequence (your own local identification code, or local ID) and a title. During the submission process, indexing staff at the database to which you are submitting will change your local ID to an accession number. You can embed other important information in the title, and Sequin uses this information to construct a record. Specifically, you can enter organism and strain or clone information in the nucleotide definition line, and gene and protein information in the protein definition line. Examples for the nucleotide and protein files are shown here:
>eIF4E [organism=Drosophila melanogaster] [strain=Oregon R] Drosophila ... CGGTTGCTTGGGTTTTATAACATCAGTCAGTGACAGGCATTTCCAGAGTTGCCCTGTTCA ... >4E-I [gene=eIF4E] [protein=eukaryotic initiation factor 4E-I] MQSDFHRMKNFANPKSMFKTSAPSTEQGRPEPPTSAAAPAEAKDVKPKEDPQETGEPAGN ... >4E-II [gene=eIF4E] [protein=eukaryotic initiation factor 4E-II] MVVLETEKTSAPSTEQGRPEPPTSAAAPAEAKDVKPKEDPQETGEPAGNTATTTAPAGDD ...
The ability to embed this information in the definition line is provided as a convenience to the submitter. If these annotations are not present, they can be entered in subsequent forms. Sequin is designed to use this information, and that provided in the initial forms, to build a properly-structured record. In many cases, the final submission can be completely prepared from these data, so that no additional manual annotation is necessary once the record is displayed.
See http://www.ncbi.nlm.nih.gov/Sequin/faq.html#Biosrcmod for a complete list of definition line modifiers.
In this example we show alternative splicing, where a single gene produces multiple messenger RNAs which encode two similar but distinct protein products.
Note that the [ and ] brackets actually appear in the text. (Brackets are sometimes used in computer documentation to denote optional text. This convention is not followed here.) The bracketed information will be removed from the definition line for each sequence. Sequin can also calculate a new definition line by computing on features in the annotated record (see "Generating the Definition Line").
Also, please note that there must be a line break (carriage return) between the definition line and the first line of sequence. Some word processors will break a single line onto two lines without actually adding a carriage return. (This feature is known as "word wrapping.") If you are unsure whether there is a carriage return, you can either set up your word processor so it shows invisible characters like carriage returns, or view the file in a text editor which does not create artificial line breaks. The definition line itself must not have a line break within it, since the second line would then be misinterpreted as the beginning of the sequence data. The actual sequence is usually broken every 50 to 80 characters, but this is not necessary for Sequin to be able to read it.
Segmented Nucleotide Sets
A segmented nucleotide entry is a set of non-contiguous sequences which has a defined order and orientation. For example, a genomic DNA segmented set could include encoding exons along with fragments of their flanking introns. An example of an mRNA segmented pair of records would be the 5' and 3' ends of an mRNA where the middle region has not been sequenced. In order to import nucleotides in a segmented set, each individual sequence must be in FASTA format with an appropriate definition line, and all sequences should be in the same file. The file containing the sequences is imported into Sequin as described later.
Population, Phylogenetic or Mutation Studies
For phylogenetic studies, the scientific name of each organism should be encoded in each FASTA definition line, e.g., [organism=Mus musculus]. For population studies, you can encode strain, clone, and isolate information in the definition line, e.g., [strain=BALB/c]. For these studies, PHYLIP, NEXUS, MACAW, and FASTA+GAP formats (see "Sample Data Files") can also be read. (All of these formats require a unique sequence identifier, i.e., a local ID.) A sample FASTA+GAP file for a phylogenetic study is shown here:
>Dmel28S [organism=Drosophila melanogaster] AUUCUGGUUAACUCUAAGCGGUGGAUCACUCGGCUCAUGGGUCGAUGAAGAACGCAGC-- AAACUGUGCGUCAUCGUGUGAACUGCAGGACACAU-GAACAUCGACAUUUUGAACGCAUA UCG-----------CAGU-------CCAUGCU-GUUAUA----------UACAACCUCAA >Xlae28S [organism=Xenopus laevis] -----UCGCGACUCUUAGCGGUGGAUCACUCGGCUCGUGCGUCGAUGAAGAACGCAGC-- UAGCUGCGAGAAUUAGUGUGAAUUGCAGA-CACAUUGAUCAUCGACACUUCGAACGCACC UUGCGGCCCCGGGUUCCUCCCGGGGCCACGCCUGUCUGAGGGUCGCUCCUCAGACCUCAG >Mmus28S [organism=Mus musculus] --------CGACUCUUAGCGGUGGAUCACUCGGCUCGUACGUCGAUGAAGAACGCAGC-- UAGCUGCGAGAAUUAAUGUGAAUUGCAGA-CACAUUGAUCAUCGACACUUCGAACGCACU U-GCGGCCCCGGGUUCCUCCCGGGGCUACGCCUGUCUGAGCGUCGCUUC-GCGACCUCAG >Hsap28S [organism=Homo sapiens] --------CGACUCUUAGCGGUGGAUCACUCGGCUCGUGCGUCGAUGAAGAACGCAGCGC UAGCUGCGAGAAUUAAUGUGAAUUGCAGA-CACAUUGAUCAUCGACACUUCGAACGCACU U-GCGGCCCCGGGUUCCUCCCGGGGCUACGCCUGUCUGAGCGUCGCUUC-GCGACCUCAG
One could add "28S ribosomal RNA gene, partial sequence" as a title to all sequences using the Annotation page of the Organism and Sequences form, and could also ask that the correct organism name be prefixed to each title. (Sequin will convert the U (uracil) characters into T (thymine) automatically.)
Sets of Segmented Sequences
If the sequences in a phylogenetic study are really segmented (e.g., ITS1 and ITS2 of ribosomal RNA without the intervening 5.8S rRNA sequence), the individual segments from a single organism can be grouped within square brackets. Subsequent segments are detected by the presence of a FASTA definition line. For example:
[ >QruITS1 [organism=Quercus rubra] CGAAAACCTGCACAGCAGAAACGACTCGCAAACTAGTAATAACTGACGGAGGACGGAGGG ... >QruITS2 CATCATTGCCCCCCATCCTTTGGTTTGGTTGGGTTGGAAGTTCACCTCCCATATGTGCCC ... ] [ >QsuITS1 [organism=Quercus suber] CAAACCTACACAGCAGAACGACTCGAGAACTGGTGACAGTTGAGGAGGGCAAGCACCTTG ... >QsuITS2 CATCGTTGCCCCCCTTCTTTGGTTTGGTTGGGTTGGAAGTTGGCCTTCCATATGTGCCCT ... ] ...
FASTA+GAP format can also use this convention for encoding sets of segmented sequences.
The sequence data we will use for this example is the genomic sequence of the Drosophila melanogaster eukaryotic initiation factors 4E-I and 4E-II (GenBank accession number U54469).
Welcome to Sequin Form
Once you have finished preparing the sequence files, you are ready to start
the Sequin program. Sequin's first window asks you to indicate the database to
which the sequence will be submitted, and prompts you to start a new project
or continue with an existing one. Once you choose a database, Sequin will
remember it in subsequent sessions. In general, each sequence submission
should be entered as a separate project. However, segmented DNA sequences,
population studies, phylogenetic studies, and mutation studies, should be
submitted together as one project. This feature also eliminates the need to
save Sequin information templates for each sequence.

See http://www.ncbi.nlm.nih.gov/Sequin/netaware.html or "Network Configuration" for instructions on how to make Sequin "network-aware". When connected to the Internet, Sequin also allows you to download a record from Entrez. If you are the original submitter of this record, you can use Sequin to edit the sequence or add further annotations to the record.
Submitting Authors Form
The pages in the Submitting Authors form ask you to provide the release date, a working title, names and contact information of submitting authors, and affiliation information. To create a personal template for use in future submissions, use the File/Export option after completing each page of this form.
The Submission page asks for a tentative title for a manuscript
describing the sequence, and will initially mark the manuscript as being
unpublished. When the article is published, the database staff will update the
sequence record with the new citation. This page also lets you indicate that a
record should be held until it is published, although the preferred policy is
to release the record immediately into the public databases.
The Contact page asks for the name, phone number, and e-mail address of the person responsible for making the submission. Database staff members will contact this person if there are any questions about the record.
The Sfx (suffix) field should be filled with personal name suffixes (e.g., Jr.,
Sr., or 3rd), not with a person's academic degrees (e.g., M.D. or Ph.D.). And
it is not necessary to type periods after initials or suffixes.
In the Authors page, enter the names of the people who should get scientific credit for the sequence presented in this record. These will become the authors for the initial (unpublished) manuscript.
Authors are entered in a spreadsheet. As soon as anything is typed in the last
row, a new (blank) row is added below it. Use the tab key to move between
fields. Tabbing from the Sfx (suffix) column automatically moves to the First
Name column in the next row. Again, suffix is for personal suffixes (e.g., Jr.
or Sr.), not academic degrees.
The Affiliation page asks for the institutional affiliation of the
primary author.
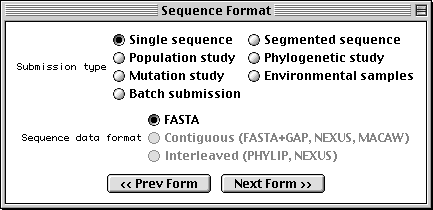
Sequence Format Form
With Sequin, the actual sequence data are imported from an outside data file. So before you begin, prepare your sequence data files using a text editor, perhaps one associated with your laboratory sequence analysis software (see "Before you Begin").
The traditional submission is of a single nucleotide sequence containing one or more genes and encoding one or more proteins. Data files should be prepared in FASTA format. Segmented sequences, e.g., where several exons have been sequenced but the complete introns are not yet sequenced, may also be submitted, with the individual nucleotide segments in FASTA format combined into one file.
Sequin will also accept population, phylogenetic, or mutation studies, and environmental samples, in which genes from many individuals or organisms are sequenced. The data for these can be entered in FASTA format, but they can also be entered in Contiguous or Interleaved formats (e.g., PHYLIP, NEXUS, MACAW, or FASTA+GAP), which contain alignment information as well as the sequences. For this example, FASTA format will be used.
The batch submission choice is provided to accommodate submission of
unrelated sequences, where no alignment is present or should be calculated.
One powerful feature of Sequin is that the program can automatically annotate the name of the organism, strain, gene, protein product, and other information on your sequence and coding regions. You can supply this information in the FASTA definition line between square brackets, e.g., [organism=Mus musculus] [strain=BALB/c] for the nucleotide and [gene=ins] [protein=insulin] for the protein.
It is much easier to produce the final submission if you let Sequin work for you in this manner.
Organism and Sequences Form
The first page of this form requests information regarding the scientific name of the organism from which the sequence was derived, if it was not already encoded in the nucleotide FASTA file. Organism information is most easily entered by selecting the appropriate organism from the scrollable list. As you begin typing the scientific name, the list will jump to the right alphabetical location. Click on the list to finish the selection.
Once you select an organism from the list, the corresponding scientific and
common names and genetic code are filled out automatically. If you then choose
Mitochondrion as the sequence location, the alternative genetic code for that
organism will be used. If your organism is not on the list, Sequin will simply
accept the scientific name you have typed; you should then manually set the
genetic code used for translation. This will later be verified by the
database staff using a more complete taxonomic database. (Sequin only holds
the top 800 organisms, while more than 40,000 are actually present in
GenBank.)
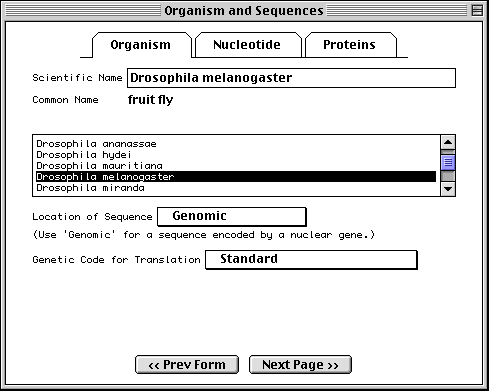
Note: For phylogenetic studies, you may annotate each FASTA definition line with [organism=scientific name] information (see "Before you Begin"), and you would not need to fill in the Organism page. When phylogenetic study has been selected as the sequence format, in fact, this page is replaced by a notice explaining how to place the organism name in the definition line. In that case you should enter the location of the sequence and the default genetic code. Similarly, in a single sequence, if you embed [organism=scientific name] in the definition line (usually along with other modifiers, such as strain or clone), you can skip this page.
Importing Nucleotide FASTA Files
To import the nucleotide sequence data, click on the Nucleotide folder tab or the Next Page button to advance to the next page. Select molecule type and topology, check any additional boxes that apply, then click on Import Nucleotide FASTA and select the appropriate file.
It is most convenient to place the multiple segments of a segmented sequence, or the individual sequences of a population, phylogenetic, or mutation study, in a single file. However, they can also be kept in separate files. In this case you would repeat the import step for each file. (In addition to importing from a file, sequences can also be read by pasting from the computer's "clipboard".)
When the sequence file import is complete, a box will appear showing the number of nucleotide segments imported, the total length in nucleotides of the sequences entered, and the local ID(s) you designated. The actual sequence data are not shown. If any of this information is missing or incorrect, check the file containing the sequence data for proper FASTA format, choose Clear from the Edit menu, then reimport the sequence(s).
The format for annotating the nucleotide FASTA definition line is shown below:
>ID [organism=scientific name] [strain=strain name] title
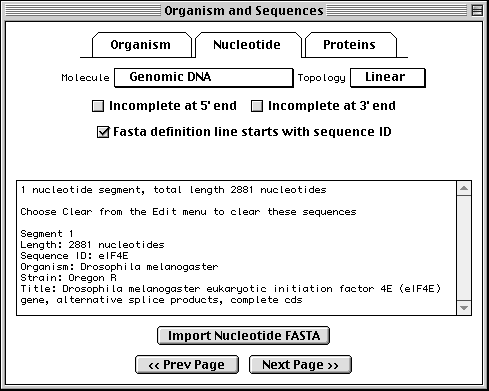
Sequin has extracted the organism and strain names from the FASTA definition line in this example, eliminating the need to fill out the Organism page.
Importing Protein FASTA Files
If you have specified a single sequence or segmented nucleotide sequence, and if it encodes one or more proteins, you can enter the sequences of the protein products in this page. To import the amino acid sequence, click on the Proteins folder tab and proceed in the same manner as for nucleotide data.
In this example, we imported two protein sequences. These are the alternative splice products of the same gene. Both protein sequences were in the same data file, but each had its own definition line with its own unique local ID.
The format for annotating the protein FASTA definition line is shown below:
>ID [gene=gene name] [protein=protein name] title
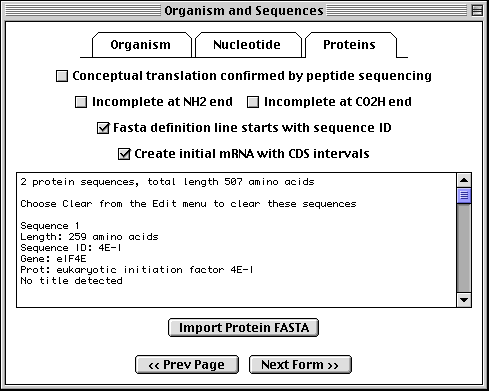
Sequin has extracted the gene and protein names from the FASTA definition lines, and will use these to construct the initial sequence record.
Annotating Population/Phylogenetic/Mutation/Environmental Sets
If you have specified a population, phylogenetic, or mutation study or
environmental sample, the
Annotation folder tab replaces the Proteins folder tab. This
page allows you to add an rRNA or CDS feature to the entire length of all
sequences in the set. In addition, you can add a title to any sequences that
didn't obtain them from a FASTA definition line. It is much easier to add
these in bulk at this step than to add individual rRNA or CDS features to each
sequence after the record is constructed.
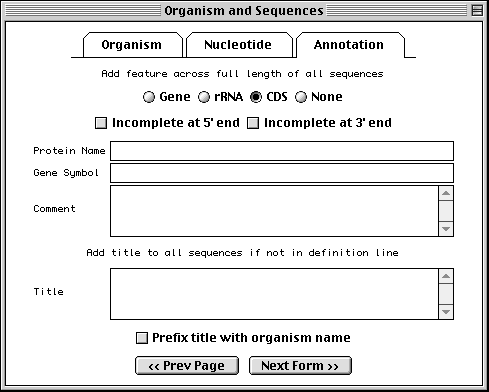
It is customary in a nucleotide record to format titles for sequences containing coding region features in the following way:
Genus species protein name (gene symbol) mRNA/gene, complete/partial cds.
The choice of "mRNA" or "gene" depends upon the molecule type (use "mRNA" for mRNA or cDNA, and "gene" for genomic DNA). Use "partial" for incomplete features. The proper organism name in a phylogenetic study can be added to the beginning of each title automatically by checking the Prefix title with organism name box.
However, for records containing with CDS, rRNA, or tRNA features, Sequin can generate the definition line automatically by computing on the features (see "Generating the Definition Line").
More complex situations, such as a population study of HIV sequences, can include multiple CDS features in each sequence. In this case, do not use the Annotation page to create features. (You can still use it for a common title, however.) After the initial submission has been created, you would manually annotate features onto one of the sequences. Feature propagation through an alignment can then be used to annotate the same features at the equivalent locations on the remaining sequences.
Entering Gene and Protein Information
In the protein FASTA definition line you can embed [gene=...] and [protein=...].
If this information was not entered in the definition line, Sequin will
display the following form for each protein sequence, allowing you to fill in
the missing information manually.
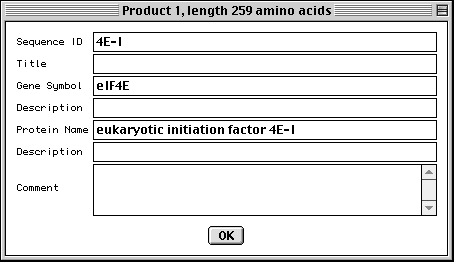
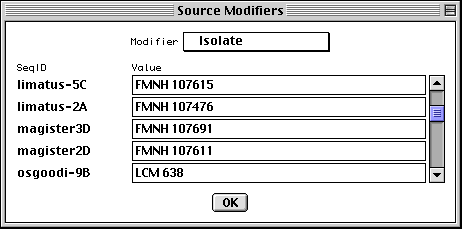
Entering Organism and Source Modifiers
In the nucleotide FASTA definition line you can embed [organism=scientific name] and modifiers from the following list (all in the format [modifier=...]): acronym, anamorph, authority, biotype, biovar, breed, cell-line, cell-type, chemovar, chromosome, clone, clone-lib, country, cultivar, dev-stage, dosage, ecotype, endogenous-virus-name, forma, forma-specialis, frequency, genotype, group, haplotype, ins-seq-name, isolate, lab-host, lineage, map, natural-host, pathovar, plasmid-name, plastid-name, pop-variant, segment, serogroup, serotype, serovar, sex, specimen-voucher, strain, sub-clone, sub-group, sub-species, sub-strain, subtype, synonym, teleomorph, tissue-lib, tissue-type, transposon-name, type, and variety. For example, [organism=Homo sapiens] [cell-line=HeLa]. Some population studies are a mixture of integrated provirus and excised virion. These can be indicated by molecule and location qualifiers. For example, [molecule=dna] [location=proviral] or [molecule=rna] [location=virion]. You can also embed [moltype=genomic] or [moltype=mRNA] to indicate from what source the molecule was isolated. If you're unsure of which modifier to use, use [note=...], and database staff will determine the appropriate modifier to use.
For population, phylogenetic, or mutation studies, Sequin presents a form to
allow this information to be edited (if it had been entered in the FASTA file),
or entered in the first place, before it constructs the sequence record. In
addition to organism name, you can enter any kind of source modifer in this
form.
GenBank View
After you have completed importing the data files, Sequin will display your
full submission information in the GenBank format (or EMBL format if you chose
EMBL as the database for submission in the first form).
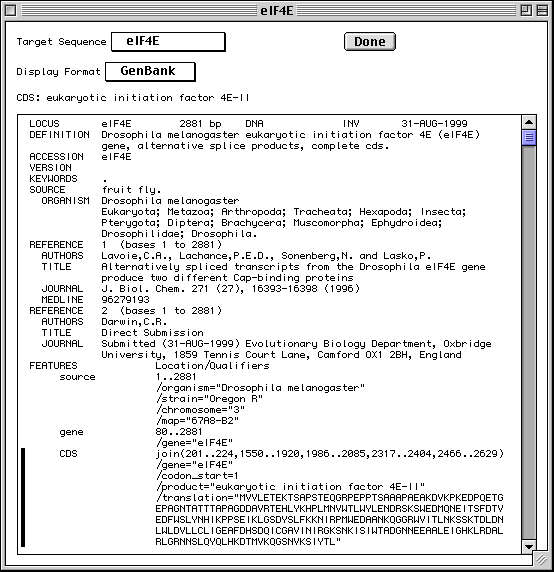
Based on information provided in your DNA and amino acid sequence files, any coding regions will be automatically identified and annotated for you. The figure shows only the top portion of the GenBank record, but you can see the first of two coding region (CDS) features. The vertical bar to the left of the paragraph indicates that the CDS has been selected by clicking with the computer's mouse.
There are also three mRNA features (not shown in the figure) that were generated by copying the cDNA feature intervals and editing them to include the 5' and 3' UTRs. Also, the journal citation, originally listed as "Unpublished", has been updated now that the article has been published. Chromosome and map information have also been added to the biological source feature. These changes were initiated by double clicking on the appropriate paragraphs in the GenBank display format. Finally, Sequin was asked to generate the definition line by computing on the annotated features.
Graphical View
To get a graphical view, use the Display Format pop-up menu to change
from GenBank to Graphic. Reviewing your submission in Graphic format allows you
to visually confirm expected location of exons, introns, and other features in
multiple interval coding regions. The Graphic view in our eukaryotic initiation
factor example illustrates how the coding region intervals for the two protein
products are spatially related to each other.
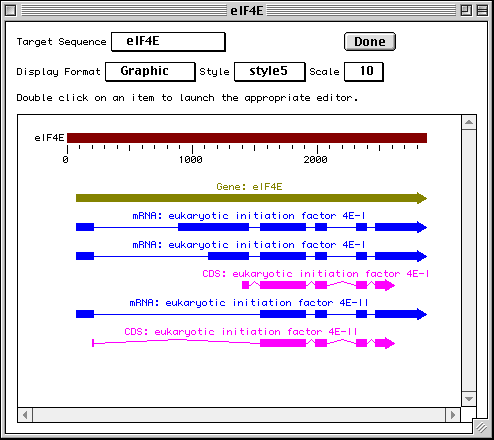
The Duplicate View item in the File menu will launch a second viewer on the record. The display format on each viewer can be independently set, allowing you to see a graphical view and a GenBank text report simultaneously. This is useful for getting an overall view of the features and seeing the details of annotation.
Sequence View
Sequence view is a static version of the sequence and alignment editor. It
shows the actual nucleotide sequence, with feature intervals annotated
directly on the sequence. Protein translations of CDS features are also shown,
as are all features shown in the graphical view. Intronic regions within
protein translations are shown as a series of tilde (~) characters.
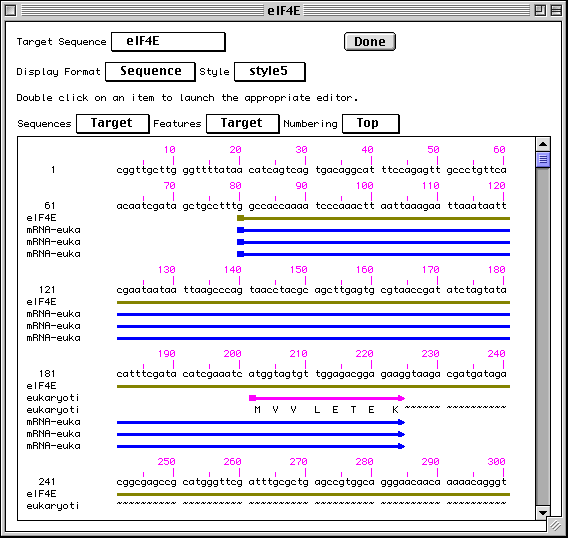
Editing and Annotating Your Submission
At this point, Sequin could process your entry based on what you have entered so far, and you could send it to your nucleotide database of choice (as set in the initial form). However, to optimize usefulness of your entry for the scientific community, you may wish to provide additional information to indicate biologically significant regions of the sequence. But first, save the entry so that if you make any unwanted changes during the editing process you can revert to the original copy.
Additional information may be in the form of Descriptors or Features. (Descriptors are annotations that apply to an entire sequence or set of sequences. Features are annotations that apply to a specific sequence interval.)
Sequin provides two convenient methods to modify your entry: (1) to edit existing information, double click on the text or graphic area you wish to modify, and Sequin will display forms requesting needed information; or (2) to add new information, use the Annotate menu and select from the list of available annotations.
Sequence Editor
Additional sequence data can also be added using Sequin's powerful sequence editor. Sequin will automatically adjust feature intervals when editing the sequence. Prior to Sequin, it was usually easier to reannotate everything from scratch when the sequence changed. But an even easier way to update sequences is described in the following section.
Updating the Sequence
Sequin can also read in a replacement sequence, or an overlapping sequence extension, and perform the alignment and feature propagation calculations necessary to adjust feature intervals, even though the individual editing operations were not done with the sequence editor.
The Update Sequence submenu within the Edit menu has several choices. These are for use by the original submitter of a record.
You can read a FASTA file or raw sequence file. This can be a replacement sequence, or it can overlap the original sequence at the 5' or 3' end. After Sequin aligns the two sequences, you then select replace or merge, as appropriate, and the sequence in your record is updated, with all feature intervals adjusted properly.
You can also update with an existing sequence record that contains features. This can be obtained from a file, or retrieved from Entrez either via an accession number or by selecting an alignment after running PowerBLAST. The latter two choices require the network-aware version of Sequin. Once it gets the new record, Sequin aligns the two sequences as before. This is typically used either to merge two records that overlap, or to copy features from database records onto a new large contig.
Generating the Definition Line
The Generate Definition Line item in the Annotate menu can make the appropriate titles once the record has been annotated with features. The general format for sequences containing coding region features is:
Genus species protein name (gene symbol) mRNA/gene, complete/partial cds.
Exceptional cases, where this automatic function is unable to generate a reasonable definition line, will be edited by the database staff to conform to the style conventions.
The new definition line will replace any previous title, including that originally on the FASTA definition line.
Validation
Once you are satisfied that you have entered all the relevant information, save
your file! Then select Validate under the Search menu. You will
either receive a message that the validation test succeeded or see a screen
listing the validation errors and warnings. Just double click on an error item
to launch the appropriate editor for making corrections. See the Sequin
Help text for more information on correcting errors. The validator
includes checks for such things as missing organism information, incorrect
coding region lengths, internal stop codons in coding regions, inconsistent
genetic codes, mismatched amino acids, and non-consensus splice sites.

Submitting the Entry
When the entry is properly formatted and error-free, click the Done button or select Prepare Submission under the File menu. You will be prompted to save your entry and e-mail it to the database you selected. The address for GenBank is gbsub@ncbi.nlm.nih.gov. The address for EMBL is datasubs@ebi.ac.uk. The address for DDBJ is ddbjsub@ddbj.nig.ac.jp.
Feature Editor Design
Sequin uses a common structure for all feature editor forms, with (usually) three top-level folder tabs. One folder tab page is specific to the given feature type (biological source and publications have more). The Properties and Location pages are common to all features. Some of these pages may have subpages, accessed by a secondary set of smaller folder tabs. This organization allows editors for complex data structures to fit in a reasonably small window size. The most important information in a given section is always presented in the first subpage.
Coding Region Page
The coding region editor is perhaps the most complicated form in Sequin. Within the Coding Region page, the Product subpage lets you predict the coding region intervals from the protein sequence or translate the protein sequence from the location. (Importing a protein sequence from a file will also interpret the [gene=...] and [protein=...] definition line information and automatically attempt to predict the coding region intervals.) It also displays the genetic code used for translation and the reading frame. (Please note that there are currently 13 different genetic codes present in Sequin. For more information on these, see http://www.ncbi.nlm.nih.gov/Taxonomy/.)
The Protein subpage lets you set the name (or, if not known, a
description) of the protein product. The Exceptions subpage allows you
to indicate translation exceptions to the normal genetic code, such as
insertion of selenocysteine, suppression of terminator codons by a suppressor
tRNA, or completion of a stop codon by poly-adenylation of an mRNA.
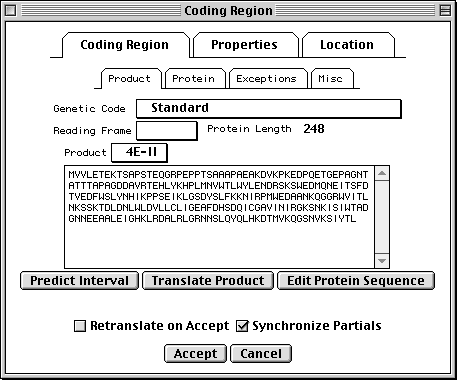
Additional annotation on the protein product might include a leader peptide, transmembrane regions, disulfide bonds, or binding sites. These can be added after setting the Target Sequence popup on the sequence viewer to the desired protein sequence. You can also launch a duplicate view, already targeted to the appropriate protein, from the Protein subpage.
Properties Page
All features have a number of fields in common. Check the Partial box to indicate that the range of the feature extends beyond the length of the sequence. (You would usually also check the 5' Partial or 3' Partial boxes in the Location page.) Exception means that the sequence of the protein product doesn't match the translation of the DNA sequence because of some known biological reason (e.g., RNA editing). Evidence can be experimental or non-experimental. For a coding region, the isolation of an mRNA or cDNA in the laboratory is sufficient to justify setting this box to Experimental.
In addition, nucleotide features (other than genes themselves) can reference a gene feature. This is frequently done by overlap. (The overlapping gene will show up on the feature as a /gene qualifier in GenBank format.) Extension of the feature location will automatically extend the gene that is selected in the editor. In rare cases, you may want to set a gene by cross-reference. For example, the mRNA for the lac operon in E. coli could indicate the lacIZYA gene by cross-reference, and the four coding regions would be overlapped by the lacI, lacZ, lacY and lacA genes.
The Comment subpage allows text to be associated with a feature. In
GenBank format, this appears as a /note qualifier. The Citations
subpage attaches citations to the feature. (The citations should first be
added to the record using items in the Publication submenu of the
Annotate menu, whereupon it will appear in the REFERENCE section.) For
example, an article that justifies a non-obvious or controversial biological
conclusion would be cited here. In GenBank format, for example, if the
publication is listed as Reference 2, the feature citation appears as
/citation=[2]. Cross-Refs are cross-references to other databases. The
contents of this subpage may only be changed by the GenBank, EMBL, or DDBJ
database staff.
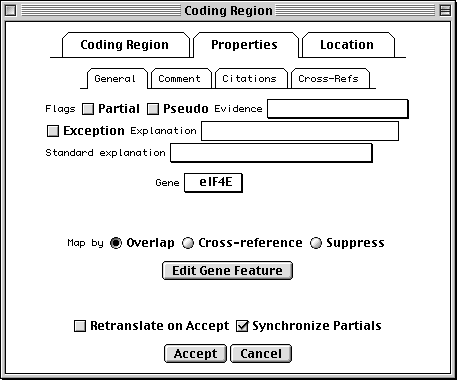
Location Page
All features are required to have a location, i.e., one or more intervals on a
sequence coordinate. The Location page provides a spreadsheet for
entering and editing this information. An arbitrary number of lines can be
entered. In this coding region example, the intervals correspond to the exons.
For an mRNA, the intervals would be the exons and UTRs. The 5'
Partial and 3' Partial check boxes will show up as < or > in
front of a feature coordinate in the GenBank flat file, indicating partial
locations.
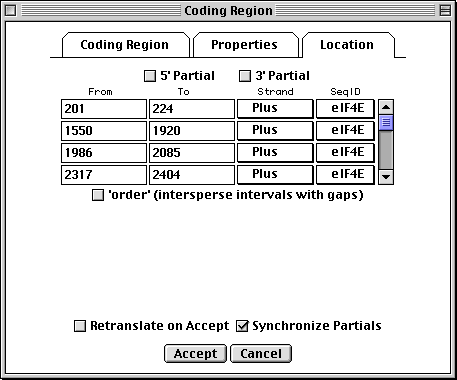
The GenBank flat file view of this location would be:
join(201..224,1550..1920,1986..2085,2317..2404,2466..2629)
If the 5' Partial or 3' Partial boxes were checked, < and > symbols would appear at the appropriate end of the join statement:
join(<201..224,1550..1920,1986..2085,2317..2404,2466..>2629)
If the sequence was reverse complemented (based on a length of 2881 nucleotides), the Strand popups would all indicate Minus, and the join statement for the resulting feature location would be as follows:
complement(join(253..416,478..565,797..896,962..1332, 2658..2681))
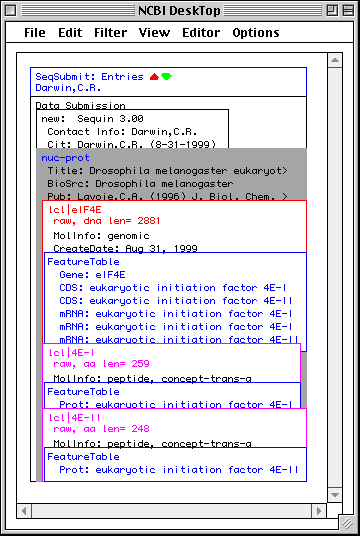
NCBI DeskTop
The NCBI DeskTop is a window that directly displays the internal structure of the record being viewed in Sequin. It can be understood as a Venn diagram. It is only meant for advanced users.
As with other views on a record, the DeskTop indicates selected items, and lets you select items by clicking. Selected items can then be the target of actions chosen from the Filter menu. Drag and drop can be used to add, rearrange, or remove items in a record.
In this example, Sequin was given the genomic nucleotide, cDNA, and protein
sequences for Drosophila eukaryotic initiation factor 4E. It then determined
the mRNA and coding region intervals, and built an initial structure. The
organism (BioSource descriptor) is at the nuc-prot set, and thus applies to
both the nucleotide and protein sequences.
Additional Information
The Sequin home page http://www.ncbi.nlm.nih.gov/Sequin/ has a Frequently Asked Questions section, and more detailed instructions on using the capabilities of network-aware Sequin.
When first downloaded, Sequin runs in stand-alone mode, without access to the network. However, the program can also be configured to exchange information with the NCBI (GenBank) over the Internet. The network-aware mode of Sequin is identical to the stand-alone mode, but it contains some additional useful options.
Sequin can only function in its network-aware mode if the computer on which it resides has a direct Internet connection. Electronic mail access to the Internet is insufficient. In general, if you can install and use a WWW-browser on your system, you should be able to install and use network-aware Sequin. Check with your system administrator or Internet provider if you are uncertain as to whether you have direct Internet connectivity.
To launch the configuration form, select Net Configure under the Misc menu,
from either the initial Welcome to Sequin form or from a viewer on an existing
sequence record.
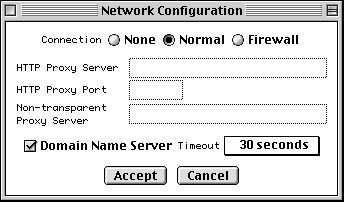
If you are not behind a firewall, set the Connection control to Normal. If you also have a Domain Name Server (DNS) available, you can now simply press Accept.
If DNS is not available, uncheck the Domain Name Server button. If you are behind a firewall, set the Connection control to Firewall. The Proxy box then becomes active. If you also use a proxy server, type in its address. (If you have DNS, it will be of the form www.myproxy.myuniversity.edu. If you do not have DNS, you should use the numerical IP address of the form 127.45.23.6.) Once you type something in the Proxy box, the Port box and Transparent Proxy button become active and can be filled in or changed as appropriate. (By default the Transparent Proxy button is off, indicating a CERN-like proxy.) Ask your network administrator for advice on the proper settings to use.
If you are in the United States, the default Timeout of 30 seconds should suffice. From foreign countries with poor Internet connection to the U.S., you can select up to 5 minutes as the timeout.
Finally, you will need to quit and restart Sequin in order for the network-aware settings to take effect.
If you are behind a firewall, it must be configured correctly to access NCBI services. Your network administrator may have done this already. If not, please have them contact NCBI for further instructions on setting up firewalls to work with NCBI services.
The following section is intended for network administrators:
Using NCBI services from behind a security firewall requires opening ports in your firewall. The ports to open are:
Firewall Port IP Address
--------------------------------
5853 130.14.22.1
5859 130.14.22.2
5840 130.14.22.8
5845 130.14.22.12
If your firewall is not transparent, the firewall port number should be mapped to the same port number on the external host.
Note: Old NCBI clients used different application configuration settings and ports than listed above. If you need to support such clients, which are being obsoleted, please contact info@ncbi.nlm.nih.gov for further information.
Complete examples of nucleotide data files in each of the supported input formats are given below.
For alignment formats, such as NEXUS, do not use the ? character to represent ambiguous bases within sequences in the alignment. This is because Sequin removes non-IUPAC characters when it imports sequences. Ambiguous bases should be indicated as IUPAC characters such as N.
NEXUS files can contain ? for "missing" at the 5' and 3' ends of sequences. PHYLIP or FASTA+GAP files should contain - rather than ? to indicate these missing sequences.
FASTA
>ABC-1 [organism=Saccharomyces cerevisiae][strain=ABC][clone=1] ATTGCGTTATGGAAATTCGAAACTGCCAAATACTATGTCACCATCATTGA TGCACCTGGACACAGAGATTTCATCAAGAACATGATCACTGGTACTT >ABC-2 [organism=Saccharomyces cerevisiae][strain=ABC][clone=2] GATATTGCTTTATGGAAATTCGAAACTGCCAAATACTATGTCACCATCAT TGATGCACCTGGACACAGAAATTTCATCAAGAACATGATCACTGGTACTT >ABC-3 [organism=Saccharomyces cerevisiae][strain=ABC][clone=3] ATTGCTTTATGGAAATTCGAAACTGCCAAATACTATGTTATGATGCACCT GGACACAGAGATTTCATCAAAAACATGATCACTGGTACTT
FASTA+GAP
>ABC-1 [organism=Saccharomyces cerevisiae][strain=ABC][clone=1] ---ATTGCGTTATGGAAATTCGAAACTGCCAAATACTATGTCACCATCAT TGATGCACCTGGACACAGAGATTTCATCAAGAACATGATCACTGGTACTT >ABC-2 [organism=Saccharomyces cerevisiae][strain=ABC][clone=2] GATATTGCTTTATGGAAATTCGAAACTGCCAAATACTATGTCACCATCAT TGATGCACCTGGACACAGAAATTTCATCAAGAACATGATCACTGGTACTT >ABC-3 [organism=Saccharomyces cerevisiae][strain=ABC][clone=3] ---ATTGCTTTATGGAAATTCGAAACTGCCAAATACTATGTTA------- TGATGCACCTGGACACAGAGATTTCATCAAAAACATGATCACTGGTACTT
PHYLIP
3 100
ABC-1 ---ATTGCGT TATGGAAATT CGAAACTGCC AAATACTATG TCACCATCAT
ABC-2 GATATTGCTT TATGGAAATT CGAAACTGCC AAATACTATG TCACCATCAT
ABC-3 ---ATTGCTT TATGGAAATT CGAAACTGCC AAATACTATG TTA-------
TGATGCACCT GGACACAGAG ATTTCATCAA GAACATGATC ACTGGTACTT
TGATGCACCT GGACACAGAA ATTTCATCAA GAACATGATC ACTGGTACTT
TGATGCACCT GGACACAGAG ATTTCATCAA AAACATGATC ACTGGTACTT
>[org=Saccharomyces cerevisiae][strain=ABC][clone=1]
>[org=Saccharomyces cerevisiae][strain=ABC][clone=2]
>[org=Saccharomyces cerevisiae][strain=ABC][clone=3]
NEXUS Interleaved
#NEXUS
begin data;
dimensions ntax=3 nchar=100;
format datatype=dna missing=? gap=- interleave ;
matrix
[ 1 50]
ABC_1 ???ATTGCGT TATGGAAATT CGAAACTGCC AAATACTATG TCACCATCAT
ABC_2 GATATTGCTT TATGGAAATT CGAAACTGCC AAATACTATG TCACCATCAT
ABC_3 ???ATTGCTT TATGGAAATT CGAAACTGCC AAATACTATG TTA-------
[ 51 100]
ABC_1 TGATGCACCT GGACACAGAG ATTTCATCAA GAACATGATC ACTGGTACTT
ABC_2 TGATGCACCT GGACACAGAA ATTTCATCAA GAACATGATC ACTGGTACTT
ABC_3 TGATGCACCT GGACACAGAG ATTTCATCAA AAACATGATC ACTGGTACTT
;
END;
begin ncbi;
sequin
>[org=Saccharomyces cerevisiae][strain=ABC][clone=1]
>[org=Saccharomyces cerevisiae][strain=ABC][clone=2]
>[org=Saccharomyces cerevisiae][strain=ABC][clone=3]
;
end;
NEXUS Contiguous
#NEXUS
begin data;
dimensions ntax=3 nchar=100;
format datatype=dna missing=? gap=- ;
matrix
ABC_1
???ATTGCGT TATGGAAATT CGAAACTGCC AAATACTATG TCACCATCAT
TGATGCACCT GGACACAGAG ATTTCATCAA GAACATGATC ACTGGTACTT
ABC_2
GATATTGCTT TATGGAAATT CGAAACTGCC AAATACTATG TCACCATCAT
TGATGCACCT GGACACAGAA ATTTCATCAA GAACATGATC ACTGGTACTT
ABC_3
???ATTGCTT TATGGAAATT CGAAACTGCC AAATACTATG TTA-------
TGATGCACCT GGACACAGAG ATTTCATCAA AAACATGATC ACTGGTACTT
;
END;
begin ncbi;
sequin
>[org=Saccharomyces cerevisiae][strain=ABC][clone=1]
>[org=Saccharomyces cerevisiae][strain=ABC][clone=2]
>[org=Saccharomyces cerevisiae][strain=ABC][clone=3]
;
end;
Sequin can now annotate features by reading in a tab-delimited table. This is most often used by genome centers that store feature interval information in relational databases or spreadsheets. For most submitters, it is usually better to supply protein sequences in FASTA format with gene and protein names embedded in the definition line.
The feature table specifies the location and type of each feature, and Sequin processes the feature intervals and translates any CDSs. The table is read in the record viewer (after the sequence has been imported) using the File-->Open menu item. The table must follow a defined format. The first line starts with >Feature, a space, and then the Sequence ID of the sequence you are annotating. In the example below, eIF4E is the Sequence ID, and it is a local identifier.
The table is composed of five columns: start, stop, feature key, qualifier key, and qualifier value. The columns are separated by tabs. The first row for any given feature has start, stop, and feature key. Additional feature intervals just have start and stop. The qualifiers follow on lines starting with three tabs.
For example, a table which looks like this:
>Features lcl|eIF4E
80 2881 gene
gene eIF4E
201 224 CDS
1550 1920
1986 2085
2317 2404
2466 2629
product eukaryotic initiation factor 4E-II
1402 1458 CDS
1550 1920
1986 2085
2317 2404
2466 2629
product eukaryotic initiation factor 4E-I
note encoded by two messenger RNAs
80 224 mRNA
1550 1920
1986 2085
2317 2404
2466 2881
product eukaryotic initiation factor 4E-II
80 224 mRNA
892 1458
1550 1920
1986 2085
2317 2404
2466 2881
product eukaryotic initiation factor 4E-I
80 224 mRNA
1129 1458
1550 1920
1986 2085
2317 2404
2466 2881
product eukaryotic initiation factor 4E-I
will result in a GenBank flatfile which contains this:
mRNA join(80..224,1129..1458,1550..1920,1986..2085,2317..2404,
2466..2881)
/gene="eIF4E"
/product="eukaryotic initiation factor 4E-I"
mRNA join(80..224,892..1458,1550..1920,1986..2085,2317..2404,
2466..2881)
/gene="eIF4E"
/product="eukaryotic initiation factor 4E-I"
mRNA join(80..224,1550..1920,1986..2085,2317..2404,2466..2881)
/gene="eIF4E"
/product="eukaryotic initiation factor 4E-II"
gene 80..2881
/gene="eIF4E"
CDS join(201..224,1550..1920,1986..2085,2317..2404,2466..2629)
/gene="eIF4E"
/codon_start=1
/product="eukaryotic initiation factor 4E-II"
/translation="MVVLETEKTSAPSTEQGRPEPPTSAAAPAEAKDVKPKEDPQETG
EPAGNTATTTAPAGDDAVRTEHLYKHPLMNVWTLWYLENDRSKSWEDMQNEITSFDTV
EDFWSLYNHIKPPSEIKLGSDYSLFKKNIRPMWEDAANKQGGRWVITLNKSSKTDLDN
LWLDVLLCLIGEAFDHSDQICGAVINIRGKSNKISIWTADGNNEEAALEIGHKLRDAL
RLGRNNSLQYQLHKDTMVKQGSNVKSIYTL"
CDS join(1402..1458,1550..1920,1986..2085,2317..2404,
2466..2629)
/gene="eIF4E"
/note="encoded by two messenger RNAs"
/codon_start=1
/product="eukaryotic initiation factor 4E-I"
/translation="MQSDFHRMKNFANPKSMFKTSAPSTEQGRPEPPTSAAAPAEAKD
VKPKEDPQETGEPAGNTATTTAPAGDDAVRTEHLYKHPLMNVWTLWYLENDRSKSWED
MQNEITSFDTVEDFWSLYNHIKPPSEIKLGSDYSLFKKNIRPMWEDAANKQGGRWVIT
LNKSSKTDLDNLWLDVLLCLIGEAFDHSDQICGAVINIRGKSNKISIWTADGNNEEAA
LEIGHKLRDALRLGRNNSLQYQLHKDTMVKQGSNVKSIYTL"
Note that if the gene feature spans the intervals of the CDS and mRNA features for that gene, you don't need to include gene "qualifiers" in those features, since they will be picked up by overlap.
Features which are on the complementary strand are indicated by reversing the interval locations. For example, the table:
>Features lcl|dna2
5284 5202 tRNA
product tRNA-Glu
will result in a GenBank flatfile containing:
tRNA complement(5202..5284)
/product="tRNA-Glu"
Questions or Comments?
Write to the NCBI Service Desk
Revised February 15, 2001