 |
|
The Materials Project

It takes an average of 20 years to bring new materials from lab to commercialization, and new materials are often discovered through a mix of intuition and serendipity. This development process makes it difficult to bring new materials needed urgently (like better Li-ion batteries, solar PV, or thermoelectrics) to market in a reasonable time.
The goal of the Materials Project is to accelerate the materials discovery and design process by (a) computing the fundamental properties of all known inorganic compounds and (b) helping researchers predict new materials with desired properties. Such information would allow researchers to rationally design new materials, dramatically cutting discovery and optimization times. The Materials Project includes aspects of computer science (software design, web design, database design), high-performance computing, theoretical physics (accurately modeling diverse chemical spaces), and materials science (linking fundamental physics to device properties).
The Materials Project infrastructure has already been used internally to discover new materials for Li-ion battery cathodes, and is now being applied to find new materials for solar photovoltaics and thermoelectrics. An online version of the database, for free use by materials researchers, is being developed in collaboration with Lawrence Berkeley Lab and is available at http://www.materialsproject.org - we hope that it accelerates your materials discovery!
|
|
|
Design and Discovery of Novel Li-Battery Electrodes
New cathode materials have the potential to increase the energy, safety, rate capability, and safety of Li ion batteries while lowering their cost. Such materials could not only find application in consumer electronics, but also help spur the clean energy revolution by making electric vehicles more affordable and capable.
We are engaged in a large-scale search for new cathode materials by performing high-throughput ab initio computations to evaluate the relevant battery properties of tens of thousands of compounds. The high-throughput battery project spans the full range from theory/computation to synthesis, characterization, and optimization. By integrating with the Materials Project framework and with novel compound prediction algorithms, the project has screened tens of thousands of candidate materials for their potential to store large amounts of energy, their capability to rapidly charge and discharge, and their stability and safety during cycling. The best candidates from these ab initio computations are then synthesized and tested by our experimental team. New materials are subsequently optimized both through experimental methods and by further computational study.
The high throughput battery project has to date predicted and synthesized three promising new cathode materials. In addition, the large data sets generated by high-throughput computation are being data-mined to learn new principles for designing future cathode materials.
|
|
|
|
Crystal Structure Prediction
Predicting unknown inorganic compounds and their crystal structure is of major importance in computational materials design. Indeed, while many properties of materials can be computed by ab initio computations, to be relevant those properties need to be computed on compounds stable enough to be synthesized. This compound and crystal structure prediction problem is most often tackled through optimization approaches, trying to find the crystalline degrees of freedom (lattice parameters and atomic positions) that will minimize the thermodynamically relevant (free) energy. The energy landscape to explore is unfortunately very rugged, with many local minima and its exploration requires advanced optimization techniques such as simulated annealing or genetic algorithms.
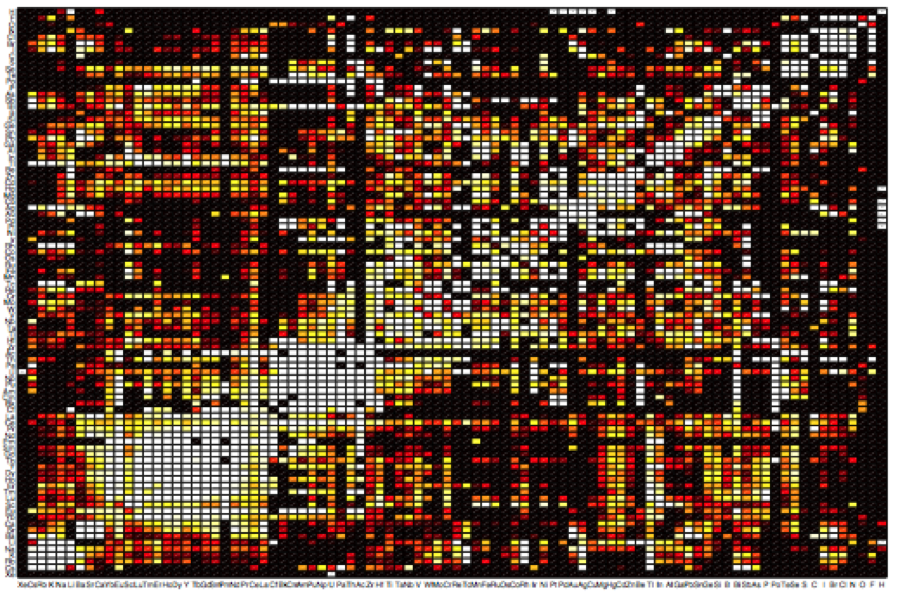
While this optimization approach is appealing, it is very computationally expensive and our research group has been pioneering alternative, cheaper compound predictions techniques based on a combination of data mining and ab initio computations. While several algorithms have been developed, the general idea is to use a database of known compounds to learn the chemical rules governing phase stability. Chemists often propose new compounds using their experience and chemical rules (ionic size, position in the periodic table etc.). Likewise, our methods perform this type of predictions using mathematical models fitted on previous knowledge.
While part of our work has been focusing on the development of tools and algorithms for data mined compound prediction, we also have been using heavily those techniques in the different high-throughput computational materials discovery projects (e.g., solar and battery projects).
|
|
Figure : Substitution map, showing how strongly an element can be substituted to another in inorganic structures.
|
|
|
Rate capabilities of Li-ion batteries : The case of LiFePO4
LiFePO 4 is an important commercial cathode material used in Li-ion batteries in high-rate applications (such as power tools and electrified vehicles) due to its high safety, reasonable capacity and high rate capabilities, provided the particle size is reduced to the nano-scale (~50-100nm).
Despite its commercial importance, the physical origin of the high rate capabilities in LiFePO4 is still poorly understood. This complication stems from the fact that bulk LiFePO4 phase-separates into an Li-rich (≈LiFePO4) and an Li-deficient (≈FePO4) phase at room temperature. Lithiation mechanisms of two-phase systems are conventionally described with nucleation and growth models, a process which involves a high kinetic barrier [Malik et al, Nature Materials, 2011]. As a result, conventional models are unable to explain the high rates observed in LiFePO4.
The goal of our research is to understand the fundamental lithiation mechanisms taking place in nano-scale LiFePO4. Such an understanding of LiFePO4 kinetics can not only shed light on a hotly debated question in the literature, but also allow us to understand the conditions under which phase-separating materials can achieve high rates. Such knowledge could open an entirely new space of potential cathode materials for Li-ion batteries.
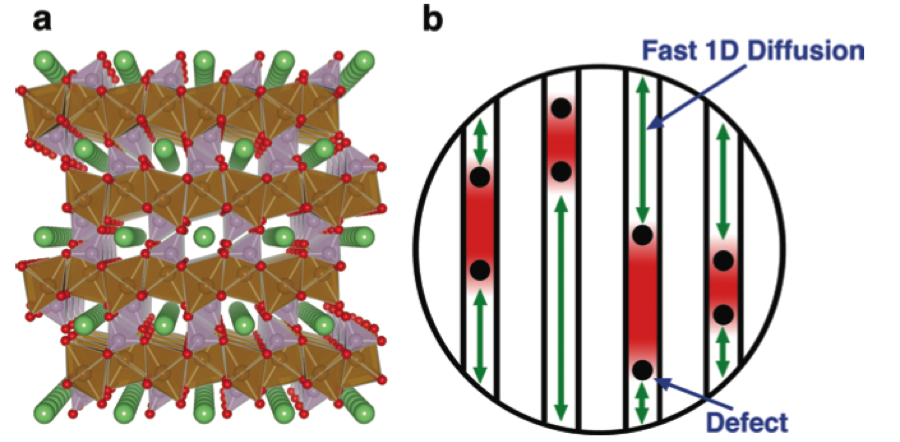
Using ab initio calculations based on Density Functional Theory (DFT), our group was the first to predict the fast one-dimensional nature of lithium diffusion in LiFePO4 [Morgan et al, Electrochemical and Solid State Letters , 2004]. Using DFT energy calculations in concert with Monte Carlo simulations, we showed the existence of non-equilibrium solid solution paths in LiFePO4, enabling fast lithiation while obviating the need for nucleation and growth.
Our fundamental understanding of the kinetics and thermodynamics of LiFePO4 has led to a novel synthesis method for LiFePO4, enabling record-breaking rate performances in this material [Kang et al, Nature, 2009]. Also, we were also able to explain why micron-sized particles exhibit much poorer rate than their nano-sized counterparts, through the increased presence of immobile channel blocking defects [Malik et al, Nano Letters, 2010].
|
|
Figure : (a) Crystal structure of LiFePO4 illustrating 1D Li+ diffusion channels oreinted along the [010] direction. (b) Schematic illustration of Li+ diffusion impeded by immobile point defects in 1D channels.
|
|
|
Solar Energy Conversion
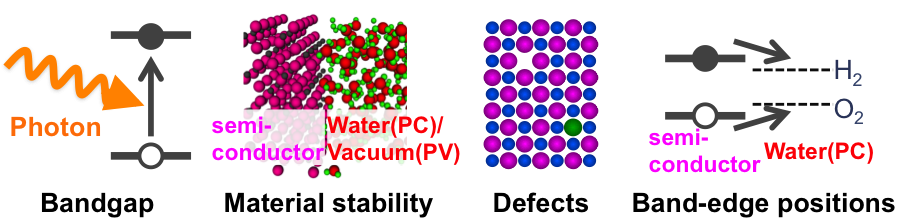
Harnessing the solar energy and converting it to more useful forms of energy, such as electrical (photovoltaics, PVs) or chemical energy (photocatalysises, PCs) has been a major ongoing global effort for decades. However, it remains a challenge to produce industrially viable, cost-effective, high-efficiency devices. A key part of this challenge is to find materials with the right properties. Some of the main issues in the materials design are material stability in operating conditions, engineering of the bandgap, adjusting band edge positions at interfaces and understanding defect mechanisms.
Our group works on computational materials design and properties prediction for inorganic semiconductors, with a focus on the above criteria. We have developed computational methodologies that enable fast and robust prediction of bandgaps [Chan et al, Physical Review Letters, 2010], band edge positions at the interface between semiconductor and water [Wu et al, Physical Review B, 2010] and material stability in water. These methods can be applied to a variety of candidate materials.
Some of our projects focus on specific materials, e.g., WO3 and pyrite (FeS2). These are currently not used in PC and PV devices, but are earth-abundant materials that are potential candidates for cheap and non-toxic solar energy conversion. We are currently investigating their key material properties, such as morphology, electronic band structure, competing phases, point defects behaviors [Sun et al, Physical Review B, 2011].
|
|
Figure: Key solar materials properties that are the object of computational research in the Ceder group. Efficient and reliable computation methods are being developed for each property, with the objective of designing better solar materials.
|
|
|
Thermoelectric Materials
Materials with the ability to directly use temperature differences to produce electrical energy, and vice-versa, have enormous potential for use in energy applications. Suggested and available applications include small scale refrigeration and climate control, cooling of photonics and microelectronics, and body heat powered personal devices.
Wide-scale industrial adoption of thermoelectrics is held back by the low efficiency of currently available materials. This presents an opportunity to develop new, higher-performing materials that better realize the potential of thermoelectric heat conversion. Recent research has suggested a path towards a new generation of thermoelectric materials via e.g., exotic bulk material compositions (the ‘phonon-glass electron-crystal’ approach) or various systems with reduced dimensionality embedded in bulk materials, such as nanoparticles in host materials or bulk nanograined composites.
Our research group is in a unique position for moving this field forward, using our strong position within energy material research, close collaboration between computational and experimental researchers, and a powerful high-throughput framework that can survey a large chemical space for potentially useful materials using state-of-the-art computational methods.
|
|