Phonon Transport
At small length scales, the classical diffusion-based model for heat conduction begins to fail. This makes modeling the energy transport challenging. In semiconductors and insulators, heat is carried primarily by vibrations in the crystal lattice known as phonons. We have been heavily involved in the development of simulation tools for multidimensional phonon transport. We have developed models based on the Boltzmann transport equation (BTE), Monte Carlo simulations, molecular dynamics (MD) simulations and also a first-principles approach based on density-functional theory to predict thermal conductivity. Some of this work is described below.
Superlattices and Thin Films

Advanced thin-film growth technology has made it possible to arrange different materials at the atomic level and to fabricate thin-film structures with strong size effects. The physical properties of these man-made structures are fundamentally different from their bulk counterparts (Figure 1).
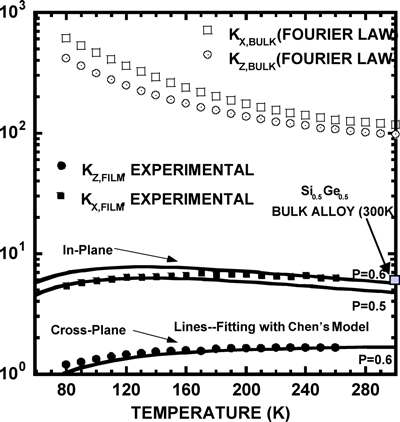
Figure 1: Click to enlarge
We conduct research on the thermal conductivity of superlattices and thin films both in-plane (parallel to the film plane) and cross-plane (in the film growth direction) directions. Various methods are used in our laboratory for thermal conductivity characterization from low to high temperatures. These include scanning probe microscopy, the 3ω method and optical diagnostics. Examples of the materials we have examined are: GaAs/AlAs, Si/Ge, InAs/AlSb, CoSb3/IrSb3 superlattices and diamond and semiconductor thin films.
In parallel with our experiments, we have used the Boltzmann transport equation, lattice dynamics, acoustic wave equations, and molecular dynamics to simulate the phonon heat conduction processes in superlattices and thin films.
Composite Structures

Figure 2: Click to enlarge
The general class of nanocomposites is a fast growing area of research. The properties of nanocomposite materials depend not only on the properties of their individual constituents but also on their morphology and interfacial characteristics. We are investigating the use of composite structures to precisely control thermal conductivity. Composites have applications in thermoelectrics, thermal management, and multifunctional materials.
We have simulated the transport of phonons through nanocomposites using both the Boltzmann transport equation and the Monte Carlo method. Figure 2 is a result from one such simulation.
Molecular Dynamics Simulations
Figure 3: Click to enlarge
As a tactic to design better thermoelectric materials with low thermal conductivity, we are using molecular dynamics (MD) simulations to investigate the phonon transport in nanostructures. MD simulations involve calculating the trajectories of particles within a system by numerically integrating classical equations of motion. From this trajectory, the system properties can be extracted as MD simulations supply detailed descriptions of the heat transfer mechanisms within nanostructures because surface, interface and impurity effects are all naturally included. In a preliminary study of a Lennard Jones crystal we analyzed the systems normal modes to learn how the modes propagate and the rate at which they decay. This information can then be used to calculate the frequency dependent contributions to the thermal conductivity, as the method can also be applied to other materials of interest.
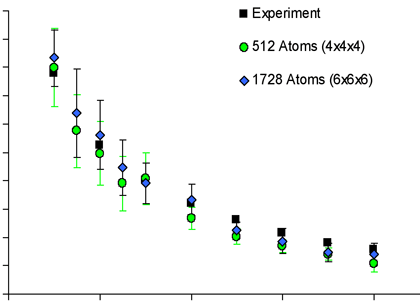
Figure 4: Click to enlarge
We plan to use MD simulations to bridge between nano and micro scale heat transfer problems, where we use the Boltzmann equation to describe the transport of phonons. The characteristics of the phonons are the essential ingredients that give materials a variety of thermal properties as we are investigating the dominant transport mechanisms in bulk silicon, as well as silicon thin films and nanowires. In addition to the Boltzmann approach we are also using the Green-Kubo formulation to directly calculate thermal conductivity, which is a very promising method that can be used to predict the thermal conductivity of nanostructures.
First-Principles Approach
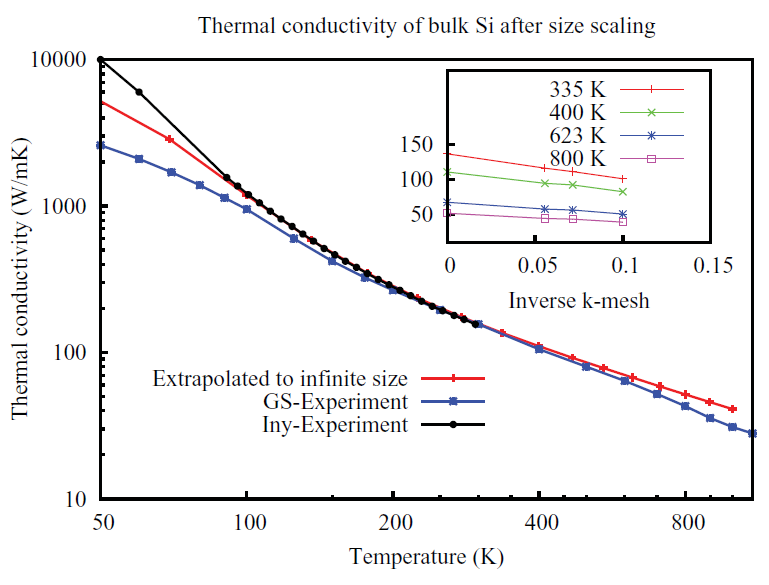
Figure 5: Click to enlarge
Selected References
- J. Shiomi, K. Esfarjani, and G. Chen, Thermal conductivity of half-Heusler compounds from first-principles calculations, Physical Review B, Vol. 84, 104302, 2011.
- K. Esfarjani, G. Chen and H. T. Stokes, Heat transport in silicon from first-principles calculations, Physical Review B, Vol. 84, 085204, 2011.
- G. Chen, "Thermal Conductivity and Ballistic Phonon Transport in Cross-Plane Direction of Superlattices," Physical Review B., Vol. 57, pp. 14958-14973
- G. Chen "Size and Interface Effects on Thermal Conductivity of Superlattices and Periodic Thin-Film Structures," ASME Journal of Heat Transfer, Vol. 119, pp. 220-229
- B. Yang and G. Chen, "Partially Coherent Phonon Heat Conduction in Superlattices," Physical Review B, Vol. 67, 195311 (1-4)
- R. Yang and G. Chen, "Thermal conductivity modeling of periodic two-dimensional nanocomposites," Phys. Rev. B 69, 195316 (2004)