FLIP VECTORS DESCRIPTION AND PROTOCOLS
All vectors will be available from Addgene in the next few weeks.
Introduction
The FLIP vectors utilize the miR30-based RNAi constructs developed for the Hannon-Elledge libraries and further improved in the lentiviral vector system pPrime (Stegmeier et al. PNAS 102(37), p. 13212). Our vectors contain the miR30-based RNAi construct after an open reading frame so expression of the upstream marker is coupled to miRNA expression.
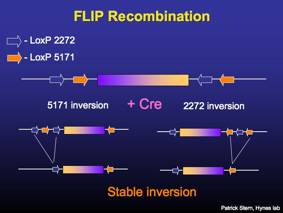
Regulated RNAi is achieved by a stable Cre-mediated recombination of a miRNA-based RNAi construct from the antisense orientation to the sense orientation. The miRNA is not processed in the antisense, but is properly processed after recombination to the sense direction.
Stable Cre-mediated inversion utilizes two different pairs of mutant loxP sites [2272 and 5171 (Siegel et al. FEBS Letters 499, p147)]. The loxP sites may mediate inversion or deletion, depending on their relative orientation to each other. The FLIP vector design is based upon a previous design pFLEX (Schnutgen et al. Nature Biotech 21, p. 562).
Vectors
All vectors are ampicillin-resistant and grown at 30C to reduce outgrowth of recombinants that may have lost portions of the vector. Recombination-suppressing bacteria (e.g. stbl3 from Invitrogen) may aid difficult cloning, but are not generally necessary. The entry vectors MSCV- and LB2-P2Gm are used for cloning and validating RNAi constructs, which are then cloned into the FLIP vectors.
Retroviral vectors are MSCV-based (MSCV2.2, Hawley) with the WPRE (Woodchuck Post- transcriptional Response Element) cloned into the ClaI site. The retroviral LTR serves as the promoter driving the expression cassette. These vectors give very good expression in murine cells (transformed cells, primary HSCs), less strong in human cells, rapidly silenced in ES cells.
Lentiviral vectors are LB2-based with two DNA insulating elements. Element #40 is before the expression cassette (5' of promoter) and SAR is after the WPRE (Kissler et al. Nature Genetics 38(4) p. 479). The basic RNAi vector contains the CAGGS (CMV-enhancer – chicken beta-actin – globin intron) promoter. The LB2- FLIP vector uses the UbiquitinC promoter, which is generally not as strong as CAGGS and may be more variable depending on cell type/ degree of transformation. With limited supporting data, UbiC is the preferred promoter for generating transgenic animals as it is less prone to heterochromatic silencing compared to CAGGS (or other viral promoters).
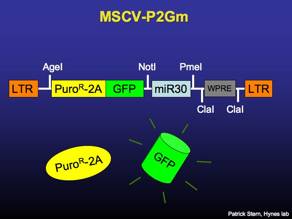
MSCV-P2Gm
MSCV-based entry vector for cloning and validating miR30-based RNAi constructs. The vector expresses puromycin-resistance and GFP, separated by the FMDV 2A peptide. [sequence link]
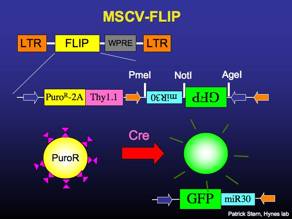
MSCV-FLIP
Constitutively expresses puromycin-resistance and the surface molecule Thy1.1. Recombines to express GFP and miR30-based RNAi. [sequence link]
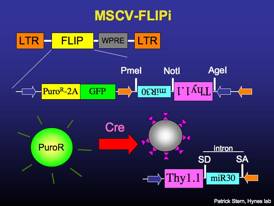
MSCV-FLIPi
Constitutively expresses puromycin-resistance and GFP. Recombines to express surface Thy1.1 and miR30-based RNAi. The miRNA is contained within an artificial intron, and splicing of the intron can boost transgene expression (1.5 to 2-fold). The expression of surface Thy1.1 facilitates antibody-mediated cell sorting. [sequence link]
LB2-CPGm
Lentiviral vector that constitutively expresses puromycin-resistance and GFP driven by the CAGGS promoter. [sequence link]
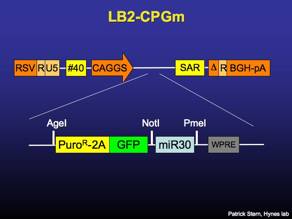
LB2-FLIP
Lentiviral vector that constitutively expresses puromycin-resistance and surface Thy1.1 driven by the UbiquitinC promoter. Recombines to express GFP and miR30-based RNAi. [sequence link]
Hairpin Cloning
The strategy for cloning shRNA sequences into the miR30-based construct is adapted from Stegmeier et al. PNAS 2005. A 97-mer oligo containing the shRNA is PCR amplified and cloned into the miR30 context in a vector. For each gene of interest, 5 or 6 knockdown constructs are usually sufficient to give 1 or 2 that reduce target gene expression >75%, but this efficiency can vary widely.
-
Design a 97-mer oligo against your gene of interest on the CSHL website. This site generates a 97-mer sequence designed for the miR30 construct. Select "psm2" and the number of oligos you want.
You will need the Accession number or sequence information.
http://katahdin.cshl.org:9331/siRNA/RNAi.cgi?type=shRNAAlternatively, you can use Biopredsi to generate 19-mer siRNA sequences. The 19-mer "guide strands" can then be run through the CSHL site to generate the appropriate 97-mers. Some have reported marginally improved knockdown efficiencies using the Biopredsi prediction algorithm.
http://www.biopredsi.org/start.html -
2. PCR amplify 97-mers with miR30 Fwd and Rev primers. These primers add XhoI and EcoRI sites for cloning into the P2Gm vectors.
miR30 Fwd:
5'-GATGGCTG-CTCGAG-AAGGTATAT-TGCTGTTGACAGTGAGCG-3'miR30 Rev:
5'-GTCTAGAG-GAATTC-CGAGGCAGTAGGCA-3'We amplify with the proof-reading polymerase Pfx or similar, though most polymerases should perform well given the short length of the template sequence. In a 50 ul reaction, amplify 100 ng template with 500 nM primers. DMSO (1-5%) may be included to facilitate template melting.
Step Temperature Time 1 94 oC 5 min 2 94 oC 30 sec 3 54 oC 30 sec 4 75 oC 30 sec 5 Repeat steps 2-4 for 16 cycles 6 75 oC 2 min 7 4 oC Forever Following amplification, purify the PCR products on a PCR purification column and elute in 50 ul EB.
-
Digest 20 ul of eluted product with XhoI and EcoRI (EcoRI buffer) overnight. The P2Gm vector may be digested overnight, or alternatively 3-4 hours is sufficient. We recommend inclusion of a phosphatase (CIP or SAP) in the vector digest to reduce background.
-
Purify digested PCR products on a PCR purification column and gel-purify the vector.
-
Ligate the hairpin into the vector at a 3:1 ratio, or approximately 4.2 ng of the hairpin insert to 100 ng of the vector, in a reaction volume of 10 uL for 1 hour at 25C.
-
Transform into competent bacteria and plate on ampicillin plates.
-
All clones must be sequence verified. Generally, we sequence 2 clones per oligo and get ~80% in the first set. We sequence from the 3' end of GFP in P2Gm with the primer below.
GFP miR sequencing:
5'-GCTGGAGTTCGTGACCGCC-3'The "empty" vector contains a hairpin targeting Firefly luciferase. Some clones may contain parental vector and the sequence is below.
Firefly hairpin
AAGGTATATTGCTGTTGACAGTGAGCGAGCTCCCGTGAATTGGAATCCTA
GTGAAGCCACAGATGTAGGATTCCAATTCAGCGGGAGCCTGCCTACTGCC
TCG
Validation
Potential shRNA sequences must be validated for gene knockdown efficacy. The P2Gm vectors confer puromycin-resistance to facilitate target cell selection. P2Gm vectors express GFP to easily determine transfection efficiency in virus-producer cells and infection of target cells. Once hairpins are validated, they are readily moved into FLIP vectors.
It is a good idea to validate knockdowns in a cell type that is closely related to the cell type you eventually intend to target. As our intended targets are generally primary cells, we generally use p53-knockdown immortalized MEFs. The p53-knockdown MEFs behave much like primary cells – contact inhibited, survive extended culture at confluency or low serum.
FLIP Cloning
The entire miR30 construct may be removed from any of these vectors by NotI-PmeI digest. A several-hour digest and a 1 hour ligation at 25C is generally sufficient for subcloning.
The gene in the antisense may be removed with AgeI-NotI. The gene and miR30 construct may be removed with AgeI-PmeI.
Transgenes that are "foreign" to the host genome and possess little homology should not present a problem when expressed in the antisense orientation. Transgenes that possess significant (antisense) homology to an element within the host mRNA may cause an "antisense effect", resulting in poor performance (interferon response and/or homologous gene/transgene destabilization).
Any transgene may possess cryptic splice sites in the antisense sequence that could result in poor virus production, due to destabilization of the proviral genome in producer cells. We have yet to encounter a gene in which this effect constrained experiments. This effect may not always affect titer but may still affect the proviral sequence (spliced provirions), so transduced cells should be examined for faithful proviral transmission.
Multiple Knockdowns in a Single Vector
The miR30 constructs may be concatamerized one after the other to give multiple shRNAs in a single vector. The strategy presented here is only for MSCV-P2Gm and not for the lenti version of P2Gm, which lacks the SwaI site.
To clone two knockdowns into a single vector, the insert miR30 is digested with NotI-PmeI. The vector containing the other miR30 is digested with NotI-SwaI. Gel purify the insert, gel or column purify the vector and ligate 1 hour at 25C.
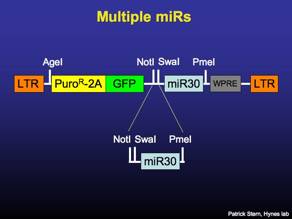
The concatamerized miR30s may now be moved with NotI-PmeI into FLIP vectors. The process can be reiterated to clone 3 miRs together.
