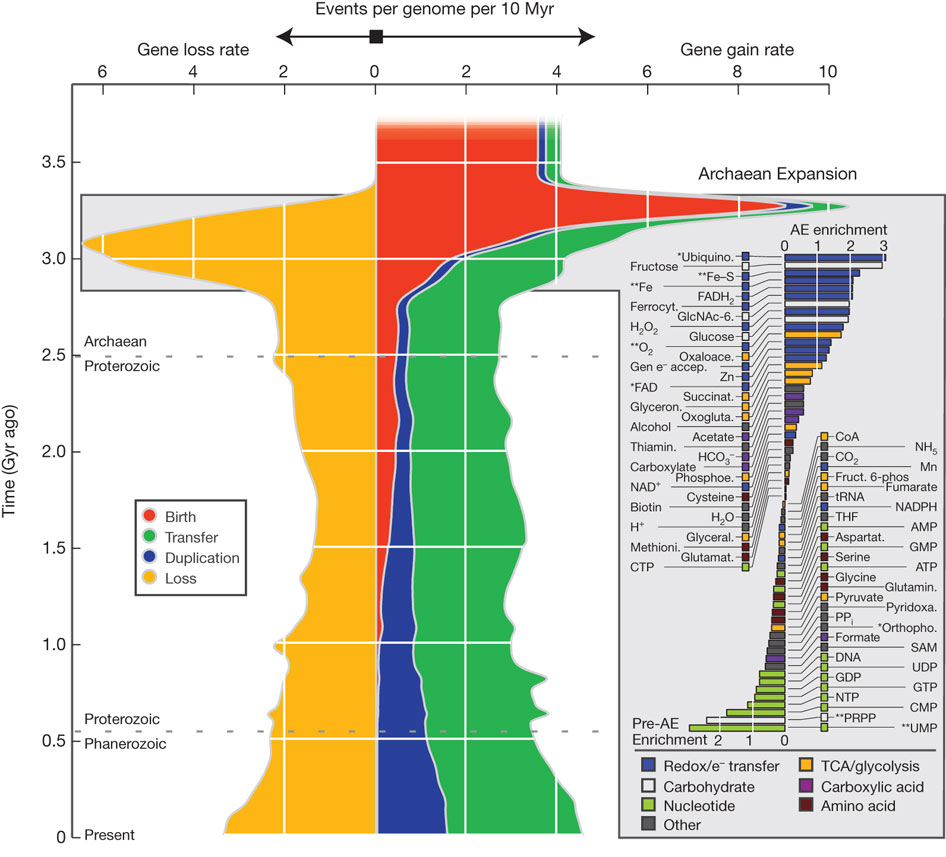
Highlights
Mobile genes in the human microbiome are structured from global to individual scalesNature 2016. doi:10.1038/nature18927.
I. L. Brito, S. Yilmaz, K. Huang, L. Xu, S. D. Jupiter, A. P. Jenkins, W. Naisilisili, M. Tamminen, C. S. Smillie, J. R. Wortman, B. W. Birren, R. J. Xavier, P. C. Blainey, A. K. Singh, D. Gevers, E. J. Alm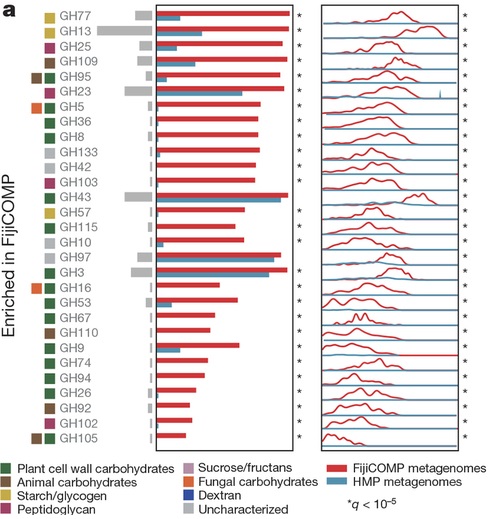
Abstract
Recent work has underscored the importance of the microbiome in human health, and has largely attributed differences in phenotype to differences in the species present among individuals. However, mobile genes can confer profoundly different phenotypes on different strains of the same species. Little is known about the function and distribution of mobile genes in the human microbiome, and in particular whether the gene pool is globally homogenous or constrained by human population structure. Here, we investigate this question by comparing the mobile genes found in the microbiomes of 81 metropolitan North Americans with those of 172 agrarian Fiji islanders using a combination of single-cell genomics and metagenomics. We find large differences in mobile gene content between the Fijian and North American microbiomes, with functional variation that mirrors known dietary differences such as the excess of plant-based starch degradation genes found in Fijian individuals. Notably, we also observed differences between the mobile gene pools of neighbouring Fijian villages, even though microbiome composition across villages is similar. Finally, we observe high rates of recombination leading to individual-specific mobile elements, suggesting that the abundance of some genes may reflect environmental selection rather than dispersal limitation. Together, these data support the hypothesis that human activities and behaviours provide selective pressures that shape mobile gene pools, and that acquisition of mobile genes is important for colonizing specific human populations.ISME J. 2016 Feb; 10(2):427-36.
Spencer SJ, Tamminen MV, Preheim SP, Guo MT, Briggs AW, Brito IL, A Weitz D, Pitknen LK, Vigneault F, Juhani Virta MP, Alm EJ.Computational and Systems Biology, Massachusetts Institute of Technology, Cambridge, MA, USA.
Abstract
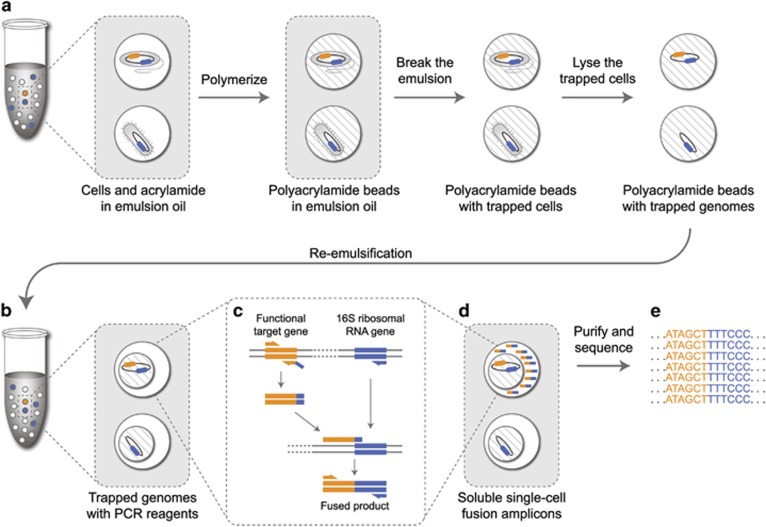
Many microbial communities are characterized by high genetic diversity. 16S ribosomal RNA sequencing can determine community members, and metagenomics can determine the functional diversity, but resolving the functional role of individual cells in high throughput remains an unsolved challenge. Here, we describe epicPCR (Emulsion, Paired Isolation and Concatenation PCR), a new technique that links functional genes and phylogenetic markers in uncultured single cells, providing a throughput of hundreds of thousands of cells with costs comparable to one genomic library preparation. We demonstrate the utility of our technique in a natural environment by profiling a sulfate-reducing community in a freshwater lake, revealing both known sulfate reducers and discovering new putative sulfate reducers. Our method is adaptable to any conserved genetic trait and translates genetic associations from diverse microbial samples into a sequencing library that answers targeted ecological questions. Potential applications include identifying functional community members, tracing horizontal gene transfer networks and mapping ecological interactions between microbial cells.Nature Biotechnology. 2015 Oct; 33(10):1053-60.
Cleary B, Brito IL, Huang K, Gevers D, Shea T, Young S, Alm EJ.Computational and Systems Biology, Massachusetts Institute of Technology, Cambridge, MA, USA.
Abstract
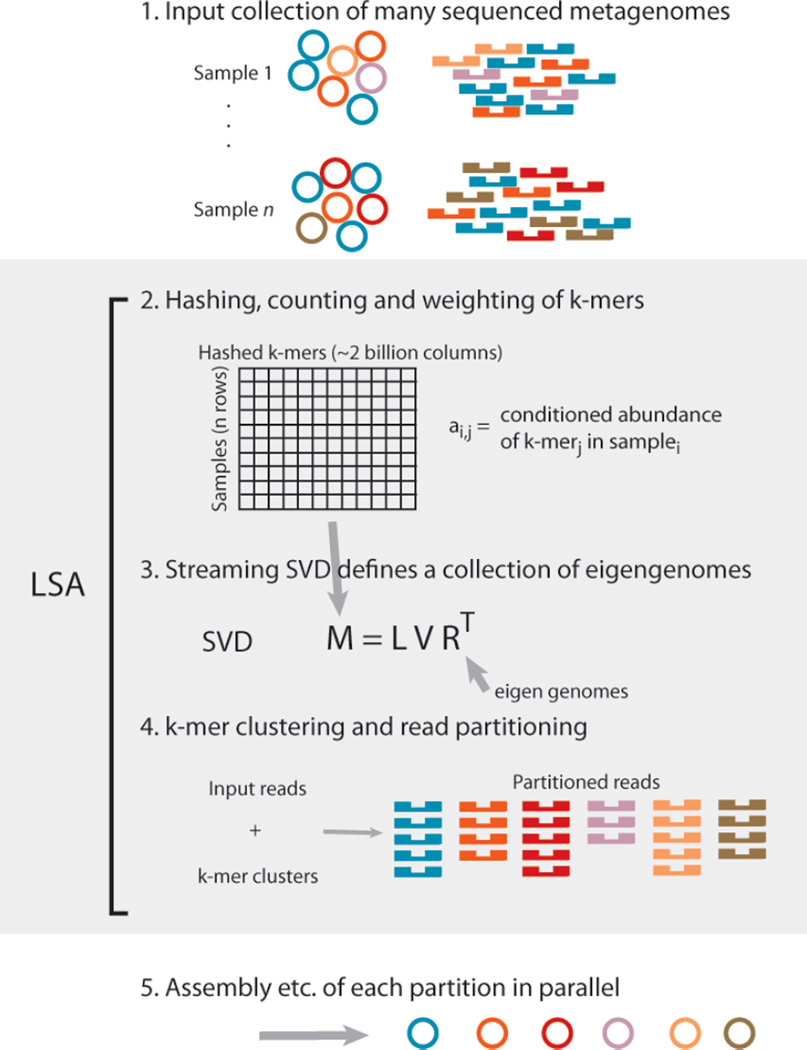
Analyses of metagenomic datasets that are sequenced to a depth of billions or trillions of bases can uncover hundreds of microbial genomes, but naive assembly of these data is computationally intensive, requiring hundreds of gigabytes to terabytes of RAM. We present latent strain analysis (LSA), a scalable, de novo pre-assembly method that separates reads into biologically informed partitions and thereby enables assembly of individual genomes. LSA is implemented with a streaming calculation of unobserved variables that we call eigengenomes. Eigengenomes reflect covariance in the abundance of short, fixed-length sequences, or k-mers. As the abundance of each genome in a sample is reflected in the abundance of each k-mer in that genome, eigengenome analysis can be used to partition reads from different genomes. This partitioning can be done in fixed memory using tens of gigabytes of RAM, which makes assembly and downstream analyses of terabytes of data feasible on commodity hardware. Using LSA, we assemble partial and near-complete genomes of bacterial taxa present at relative abundances as low as 0.00001%. We also show that LSA is sensitive enough to separate reads from several strains of the same species.PNAS. 2014 Nov 11; 111(45):16112-7.
 Oren Y, Smith MB, Johns NI, Kaplan Zeevi M, Biran D, Ron EZ, Corander J,
Wang HH, Alm EJ, Pupko T.
Oren Y, Smith MB, Johns NI, Kaplan Zeevi M, Biran D, Ron EZ, Corander J,
Wang HH, Alm EJ, Pupko T.
Abstract
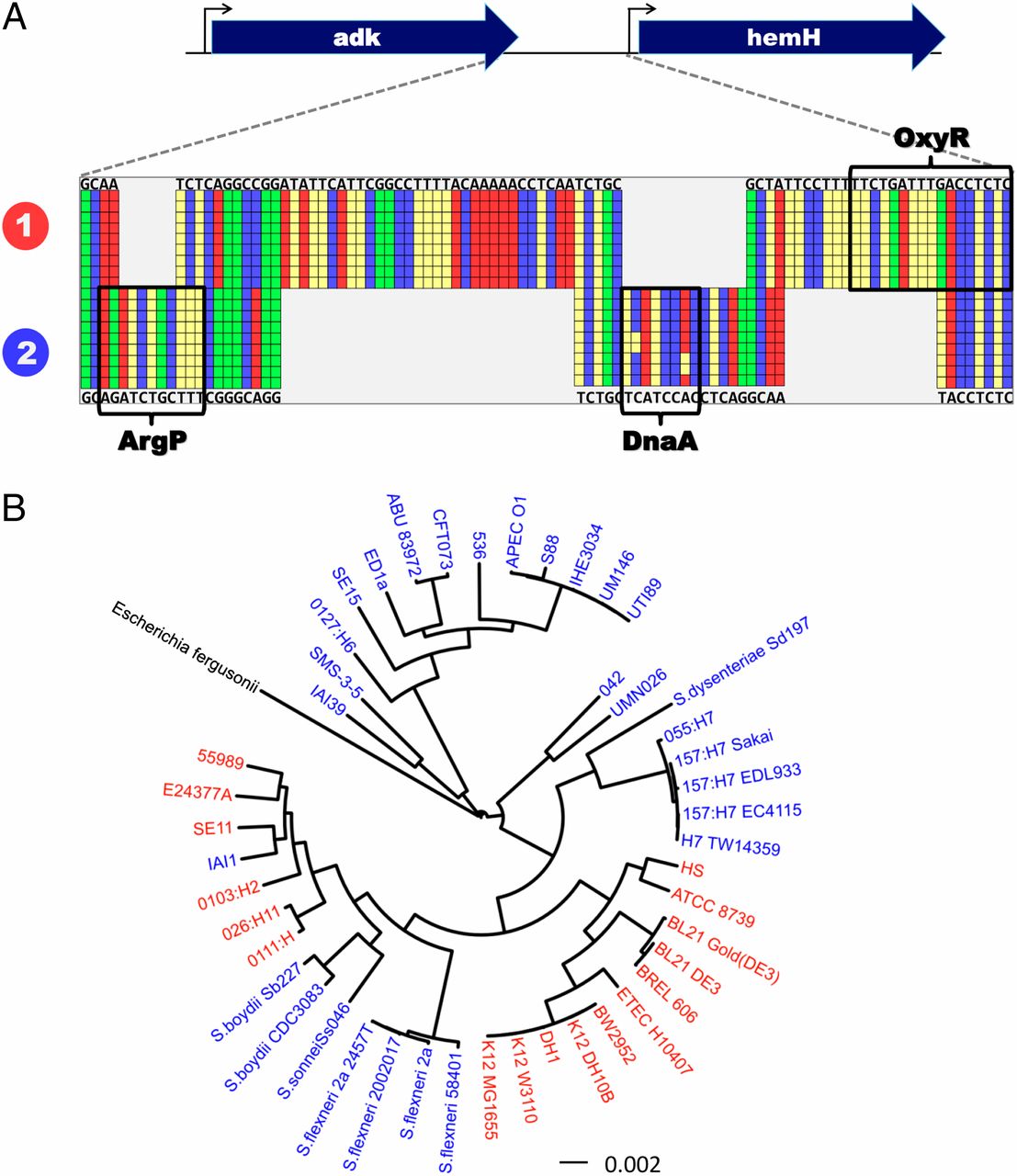
Understanding the mechanisms that generate variation is a common pursuit unifying the life sciences. Bacteria represent an especially striking puzzle, because closely related strains possess radically different metabolic and ecological capabilities. Differences in protein repertoire arising from gene transfer are currently considered the primary mechanism underlying phenotypic plasticity in bacteria. Although bacterial coding plasticity has been extensively studied in previous decades, little is known about the role that regulatory plasticity plays in bacterial evolution. Here, we show that bacterial genes can rapidly shift between multiple regulatory modes by acquiring functionally divergent nonhomologous promoter regions. Through analysis of 270,000 regulatory regions across 247 genomes, we demonstrate that regulatory "switching" to nonhomologous alternatives is ubiquitous, occurring across the bacterial domain. Using comparative transcriptomics, we show that at least 16% of the expression divergence between Escherichia coli strains can be explained by this regulatory switching. Further, using an oligonucleotide regulatory library, we establish that switching affects bacterial promoter architecture. We provide evidence that regulatory switching can occur through horizontal regulatory transfer, which allows regulatory regions to move across strains, and even genera, independently from the genes they regulate. Finally, by experimentally characterizing the fitness effect of a regulatory transfer on a pathogenic E. coli strain, we demonstrate that regulatory switching elicits important phenotypic consequences. Taken together, our findings expose previously unappreciated regulatory plasticity in bacteria and provide a gateway for understanding bacterial phenotypic variation and adaptation.Nature. 2014 Feb 19
For medical use, human stool should be considered a tissue, not a drug, argue Mark B Smith, Colleen Kelly and Eric J Alm.
Related links:
OpenBiome.org
In New York Times
Genome Biol. 2014;15(7):R89.
David LA, Materna AC, Friedman J, Campos-Baptista MI, Blackburn MC, Perrotta A, Erdman SE, Alm EJ.Computational and Systems Biology Initiative, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. .
Abstract
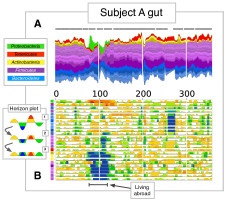
Disturbance to human microbiota may underlie several pathologies. Here, we link over 10,000 longitudinal measurements of human wellness and action to the daily gut and salivary microbiota dynamics of two individuals over the course of one year. These time series show overall microbial communities to be stable for months. However, rare events in each subjects life rapidly and broadly impacted microbiota dynamics. Travel from the developed to the developing world in one subject led to a nearly two-fold increase in the Bacteroidetes to Firmicutes ratio, which reversed upon return. Enteric infection in the other subject resulted in the permanent decline of most gut bacterial taxa, which were replaced by genetically similar species. Still, even during periods of overall community stability, the dynamics of select microbial taxa could be associated with specific host behaviors. Most prominently, changes in host fiber intake positively correlated with next-day abundance changes among 15% of gut microbiota members. Our findings suggest that although human-associated microbial communities are generally stable, they can be quickly and profoundly altered by common human actions and experiences.Publication related
Metadata for EBI Study: ERP006059Science. 2012 Apr 6;336(6077):48-51.
 Shapiro BJ, Friedman J, Cordero OX, Preheim SP, Timberlake SC, Szab G, Polz MF,
Alm EJ.
Shapiro BJ, Friedman J, Cordero OX, Preheim SP, Timberlake SC, Szab G, Polz MF,
Alm EJ.Program in Computational and Systems Biology, Massachusetts Institute of Technology, Cambridge, MA 02139, USA.
Abstract
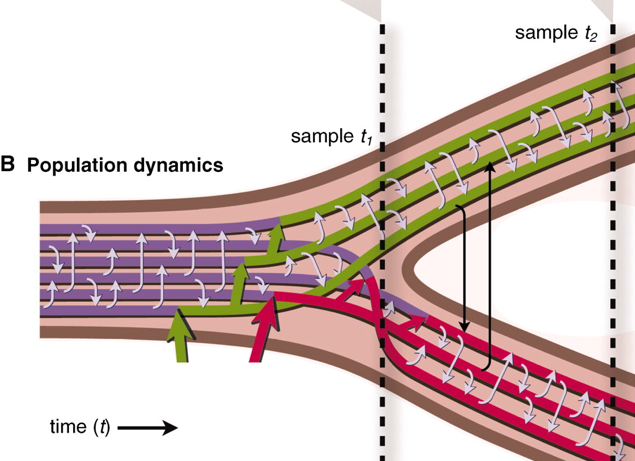
Genetic exchange is common among bacteria, but its effect on population diversity during ecological differentiation remains controversial. A fundamental question is whether advantageous mutations lead to selection of clonal genomes or, as in sexual eukaryotes, sweep through populations on their own. Here, we show that in two recently diverged populations of ocean bacteria, ecological differentiation has occurred akin to a sexual mechanism: A few genome regions have swept through subpopulations in a habitat-specific manner, accompanied by gradual separation of gene pools as evidenced by increased habitat specificity of the most recent recombinations. These findings reconcile previous, seemingly contradictory empirical observations of the genetic structure of bacterial populations and point to a more unified process of differentiation in bacteria and sexual eukaryotes than previously thought.Nature. 2011 Oct 30;480(7376):241-4.
Chris S Smillie; Mark B Smith; Jonathan Friedman; Otto X Cordero; Lawrence A David; Eric J AlmComputational and Systems Biology Initiative, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. .
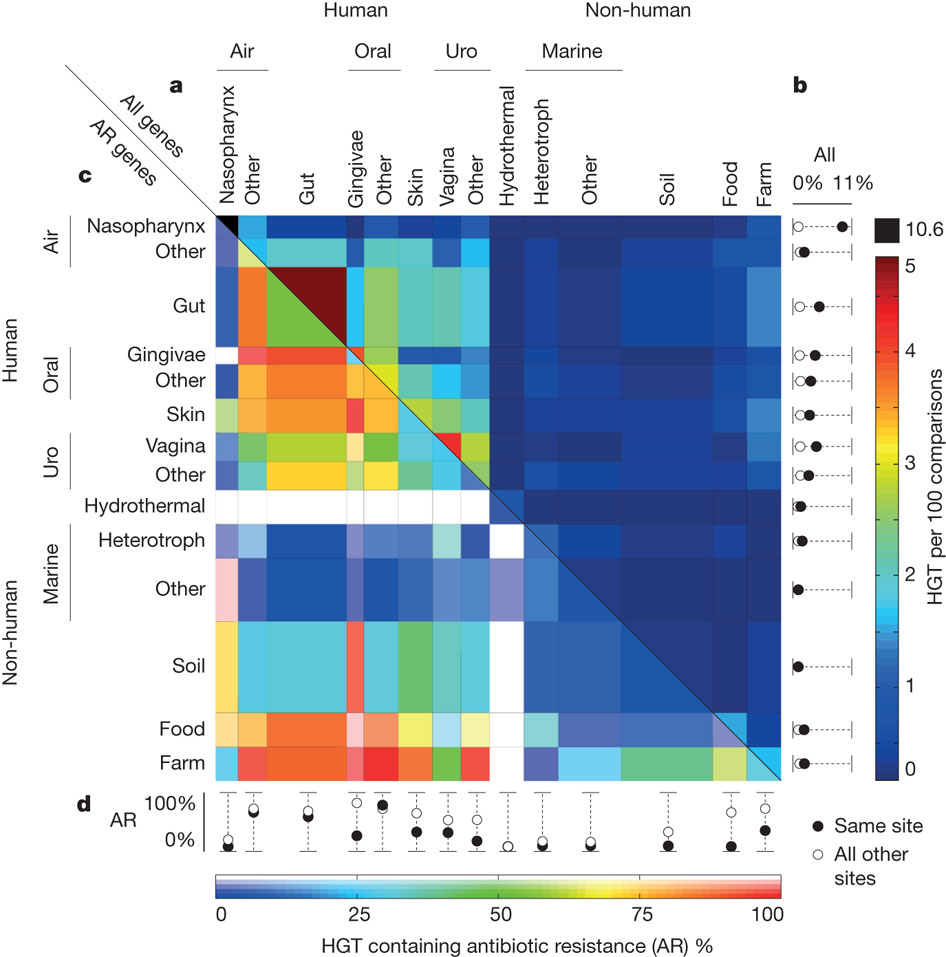
Abstract
Horizontal gene transfer (HGT), the acquisition of genetic material from non-parental lineages, is known to be important in bacterial evolution. In particular, HGT provides rapid access to genetic innovations, allowing traits such as virulence, antibiotic resistance and xenobiotic metabolism to spread through the human microbiome. Recent anecdotal studies providing snapshots of active gene flow on the human body have highlighted the need to determine the frequency of such recent transfers and the forces that govern these events. Here we report the discovery and characterization of a vast, human-associated network of gene exchange, large enough to directly compare the principal forces shaping HGT. We show that this network of 10,770 unique, recently transferred (more than 99% nucleotide identity) genes found in 2,235 full bacterial genomes, is shaped principally by ecology rather than geography or phylogeny, with most gene exchange occurring between isolates from ecologically similar, but geographically separated, environments. For example, we observe 25-fold more HGT between human-associated bacteria than among ecologically diverse non-human isolates (P = 3.0 x 10(-270)). We show that within the human microbiome this ecological architecture continues across multiple spatial scales, functional classes and ecological niches with transfer further enriched among bacteria that inhabit the same body site, have the same oxygen tolerance or have the same ability to cause disease. This structure offers a window into the molecular traits that define ecological niches, insight that we use to uncover sources of antibiotic resistance and identify genes associated with the pathology of meningitis and other diseases.Nature. 2011 Jan 6;469(7328):93-6.
David LA, Alm EJ.Computational and Systems Biology Initiative, Massachusetts Institute of Technology, Cambridge, Massachusetts 02139, USA. .