IdealMolalSoln Class Reference
[Thermodynamic Properties]
This phase is based upon the mixing-rule assumption that all molality-based activity coefficients are equal to one. More...
#include <IdealMolalSoln.h>
Public Member Functions | |
| IdealMolalSoln () | |
| Constructors. | |
| IdealMolalSoln (const IdealMolalSoln &) | |
| Copy Constructor. | |
| IdealMolalSoln & | operator= (const IdealMolalSoln &) |
| Assignment operator. | |
| IdealMolalSoln (std::string inputFile, std::string id="") | |
| Constructor for phase initialization. | |
| IdealMolalSoln (XML_Node &phaseRef, std::string id="") | |
| Constructor for phase initialization. | |
| virtual | ~IdealMolalSoln () |
| Destructor. | |
| ThermoPhase * | duplMyselfAsThermoPhase () const |
| Duplication function. | |
| virtual void | setParameters (int n, doublereal *const c) |
| virtual void | getParameters (int &n, doublereal *const c) const |
| virtual void | setParametersFromXML (const XML_Node &eosdata) |
| SpeciesThermo & | speciesThermo () |
| virtual void | initThermo () |
| Initialization routine for an IdealMolalSoln phase. | |
| void | constructPhaseFile (std::string infile, std::string id="") |
| Import and initialize an IdealMolalSoln phase specification in an XML tree into the current object. | |
| void | constructPhaseXML (XML_Node &phaseNode, std::string id) |
| Import and initialize an IdealMolalSoln phase specification in an XML tree into the current object. | |
| virtual void | initThermoXML (XML_Node &phaseNode, std::string id="") |
| Import and initialize an IdealMolalSoln phase specification in an XML tree into the current object. | |
| double | speciesMolarVolume (int k) const |
| Report the molar volume of species k. | |
| void | getSpeciesMolarVolumes (double *smv) const |
| virtual void | setStateFromXML (const XML_Node &state) |
| Set equation of state parameter values from XML entries. | |
| void | setState_TPM (doublereal t, doublereal p, const doublereal *const molalities) |
| Set the temperature (K), pressure (Pa), and molalities (gmol kg-1) of the solutes. | |
| void | setState_TPM (doublereal t, doublereal p, compositionMap &m) |
| Set the temperature (K), pressure (Pa), and molalities. | |
| void | setState_TPM (doublereal t, doublereal p, const std::string &m) |
| Set the temperature (K), pressure (Pa), and molalities. | |
| virtual std::string | report (bool show_thermo=true) const |
| returns a summary of the state of the phase as a string | |
| doublereal | _RT () const |
| Return the Gas Constant multiplied by the current temperature. | |
| bool | chargeNeutralityNecessary () const |
| Returns the chargeNeutralityNecessity boolean. | |
| XML_Node & | xml () |
| Returns a reference to the XML_Node storred for the phase. | |
| std::string | id () const |
| Return the string id for the phase. | |
| void | setID (std::string id) |
| Set the string id for the phase. | |
| std::string | name () const |
| Return the name of the phase. | |
| void | setName (std::string nm) |
| Sets the string name for the phase. | |
| void | saveState (vector_fp &state) const |
| Save the current internal state of the phase. | |
| void | saveState (int lenstate, doublereal *state) const |
| Write to array 'state' the current internal state. | |
| void | restoreState (const vector_fp &state) |
| Restore a state saved on a previous call to saveState. | |
| void | restoreState (int lenstate, const doublereal *state) |
| Restore the state of the phase from a previously saved state vector. | |
| void | setMoleFractionsByName (compositionMap &xMap) |
| Set the species mole fractions by name. | |
| void | setMoleFractionsByName (const std::string &x) |
| Set the mole fractions of a group of species by name. | |
| void | setMassFractionsByName (compositionMap &yMap) |
| Set the species mass fractions by name. | |
| void | setMassFractionsByName (const std::string &x) |
| Set the species mass fractions by name. | |
| void | setState_TRX (doublereal t, doublereal dens, const doublereal *x) |
| Set the internally storred temperature (K), density, and mole fractions. | |
| void | setState_TRX (doublereal t, doublereal dens, compositionMap &x) |
| Set the internally storred temperature (K), density, and mole fractions. | |
| void | setState_TRY (doublereal t, doublereal dens, const doublereal *y) |
| Set the internally storred temperature (K), density, and mass fractions. | |
| void | setState_TRY (doublereal t, doublereal dens, compositionMap &y) |
| Set the internally storred temperature (K), density, and mass fractions. | |
| void | setState_TNX (doublereal t, doublereal n, const doublereal *x) |
| Set the internally storred temperature (K), molar density (kmol/m^3), and mole fractions. | |
| void | setState_TR (doublereal t, doublereal rho) |
| Set the internally storred temperature (K) and density (kg/m^3). | |
| void | setState_TX (doublereal t, doublereal *x) |
| Set the internally storred temperature (K) and mole fractions. | |
| void | setState_TY (doublereal t, doublereal *y) |
| Set the internally storred temperature (K) and mass fractions. | |
| void | setState_RX (doublereal rho, doublereal *x) |
| Set the density (kg/m^3) and mole fractions. | |
| void | setState_RY (doublereal rho, doublereal *y) |
| Set the density (kg/m^3) and mass fractions. | |
| void | getMolecularWeights (vector_fp &weights) const |
| Copy the vector of molecular weights into vector weights. | |
| void | getMolecularWeights (int iwt, doublereal *weights) const |
| Copy the vector of molecular weights into array weights. | |
| void | getMolecularWeights (doublereal *weights) const |
| Copy the vector of molecular weights into array weights. | |
| const array_fp & | molecularWeights () const |
| Return a const reference to the internal vector of molecular weights. | |
| void | getMoleFractionsByName (compositionMap &x) const |
| Get the mole fractions by name. | |
| doublereal | moleFraction (int k) const |
| Return the mole fraction of a single species. | |
| doublereal | moleFraction (std::string name) const |
| Return the mole fraction of a single species. | |
| doublereal | massFraction (int k) const |
| Return the mass fraction of a single species. | |
| doublereal | massFraction (std::string name) const |
| Return the mass fraction of a single species. | |
| doublereal | chargeDensity () const |
| Charge density [C/m^3]. | |
| int | nDim () const |
| Returns the number of spatial dimensions (1, 2, or 3). | |
| void | setNDim (int ndim) |
| Set the number of spatial dimensions (1, 2, or 3). | |
| virtual void | freezeSpecies () |
| Finished adding species, prepare to use them for calculation of mixture properties. | |
| virtual bool | ready () const |
| True if both elements and species have been frozen. | |
| int | nSpecies () const |
| Returns the number of species in the phase. | |
| doublereal | molecularWeight (int k) const |
Molecular weight of species k. | |
| doublereal | molarMass (int k) const |
Return the Molar mass of species k. | |
| doublereal | charge (int k) const |
| doublereal | nAtoms (int k, int m) const |
Number of atoms of element m in species k. | |
| void | getAtoms (int k, double *atomArray) const |
| Get a vector containing the atomic composition of species k. | |
| void | stateMFChangeCalc (bool forceChange=false) |
| Every time the mole fractions have changed, this routine will increment the stateMFNumber. | |
| int | stateMFNumber () const |
| Return the state number. | |
Utilities | |
| virtual int | eosType () const |
| Equation of state type flag. | |
Molar Thermodynamic Properties of the Solution --------------- | |
| virtual doublereal | enthalpy_mole () const |
| Molar enthalpy of the solution. Units: J/kmol. | |
| virtual doublereal | intEnergy_mole () const |
| Molar internal energy of the solution: Units: J/kmol. | |
| virtual doublereal | entropy_mole () const |
| Molar entropy of the solution. Units: J/kmol/K. | |
| virtual doublereal | gibbs_mole () const |
| Molar Gibbs function for the solution: Units J/kmol. | |
| virtual doublereal | cp_mole () const |
| Molar heat capacity of the solution at constant pressure. Units: J/kmol/K. | |
| virtual doublereal | cv_mole () const |
| Molar heat capacity of the solution at constant volume. Units: J/kmol/K. | |
Potential Energy | |
| virtual void | setPotentialEnergy (int k, doublereal pe) |
| Set the potential energy of species k to pe. | |
| virtual doublereal | potentialEnergy (int k) const |
| void | setElectricPotential (doublereal v) |
| Set the electric potential of this phase (V). | |
| doublereal | electricPotential () const |
| Returns the electric potential of this phase (V). | |
Activities and Activity Concentrations | |
| virtual void | getActivityConcentrations (doublereal *c) const |
| virtual doublereal | standardConcentration (int k=0) const |
The standard concentration 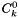 used to normalize the generalized concentration. used to normalize the generalized concentration. | |
| virtual doublereal | logStandardConc (int k=0) const |
| virtual void | getUnitsStandardConc (double *uA, int k=0, int sizeUA=6) const |
| virtual void | getActivities (doublereal *ac) const |
| virtual void | getMolalityActivityCoefficients (doublereal *acMolality) const |
Partial Molar Properties of the Solution ----------------- | |
| virtual void | getChemPotentials (doublereal *mu) const |
| Get the species chemical potentials: Units: J/kmol. | |
| virtual void | getPartialMolarEnthalpies (doublereal *hbar) const |
| Returns an array of partial molar enthalpies for the species in the mixture. | |
| virtual void | getPartialMolarEntropies (doublereal *sbar) const |
| Returns an array of partial molar entropies of the species in the solution. Units: J/kmol. | |
| virtual void | getPartialMolarVolumes (doublereal *vbar) const |
| virtual void | getPartialMolarCp (doublereal *cpbar) const |
| Partial molar heat capacity of the solution:. UnitsL J/kmol/K. | |
Chemical Equilibrium | |
| virtual void | setToEquilState (const doublereal *lambda_RT) |
| This method is used by the ChemEquil equilibrium solver. | |
Critical state properties. | |
| virtual doublereal | critTemperature () const |
| Critical temperature (K). | |
| virtual doublereal | critPressure () const |
| Critical pressure (Pa). | |
| virtual doublereal | critDensity () const |
| Critical density (kg/m3). | |
Utilities | |
| void | setpHScale (const int pHscaleType) |
| Set the pH scale, which determines the scale for single-ion activity coefficients. | |
| int | pHScale () const |
| Reports the pH scale, which determines the scale for single-ion activity coefficients. | |
Utilities for Solvent ID and Molality | |
| void | setSolvent (int k) |
| This routine sets the index number of the solvent for the phase. | |
| void | setMoleFSolventMin (doublereal xmolSolventMIN) |
| Sets the minimum mole fraction in the molality formulation. | |
| int | solventIndex () const |
| Returns the solvent index. | |
| doublereal | moleFSolventMin () const |
| Returns the minimum mole fraction in the molality formulation. | |
| void | calcMolalities () const |
| Calculates the molality of all species and stores the result internally. | |
| void | getMolalities (doublereal *const molal) const |
| This function will return the molalities of the species. | |
| void | setMolalities (const doublereal *const molal) |
| Set the molalities of the solutes in a phase. | |
| void | setMolalitiesByName (compositionMap &xMap) |
| Set the molalities of a phase. | |
| void | setMolalitiesByName (const std::string &name) |
| Set the molalities of a phase. | |
Activities, Standard States, and Activity Concentrations | |
| int | activityConvention () const |
| This method returns the activity convention. | |
| void | getActivityCoefficients (doublereal *ac) const |
| Get the array of non-dimensional activity coefficients at the current solution temperature, pressure, and solution concentration. | |
| virtual double | osmoticCoefficient () const |
| Calculate the osmotic coefficient. | |
Partial Molar Properties of the Solution | |
| void | getElectrochemPotentials (doublereal *mu) const |
| Get the species electrochemical potentials. | |
Utilities (VPStandardStateTP) | |
| virtual int | standardStateConvention () const |
| This method returns the convention used in specification of the standard state, of which there are currently two, temperature based, and variable pressure based. | |
| virtual void | getdlnActCoeffdlnC (doublereal *dlnActCoeffdlnC) const |
| Get the array of log concentration-like derivatives of the log activity coefficients. | |
Partial Molar Properties of the Solution (VPStandardStateTP) | |
| void | getChemPotentials_RT (doublereal *mu) const |
| Get the array of non-dimensional species chemical potentials These are partial molar Gibbs free energies. | |
Initialization Methods - For Internal use (VPStandardState) | |
| void | setVPSSMgr (VPSSMgr *vp_ptr) |
| set the VPSS Mgr | |
| VPSSMgr * | provideVPSSMgr () |
| Return a pointer to the VPSSMgr for this phase. | |
| void | createInstallPDSS (int k, const XML_Node &s, const XML_Node *phaseNode_ptr) |
| PDSS * | providePDSS (int k) |
| const PDSS * | providePDSS (int k) const |
Information Methods | |
| doublereal | refPressure () const |
| Returns the reference pressure in Pa. | |
| doublereal | minTemp (int k=-1) const |
| Minimum temperature for which the thermodynamic data for the species or phase are valid. | |
| doublereal | Hf298SS (const int k) const |
| Report the 298 K Heat of Formation of the standard state of one species (J kmol-1). | |
| virtual void | modifyOneHf298SS (const int k, const doublereal Hf298New) |
| Modify the value of the 298 K Heat of Formation of one species in the phase (J kmol-1). | |
| doublereal | maxTemp (int k=-1) const |
| Maximum temperature for which the thermodynamic data for the species are valid. | |
Mechanical Properties | |
| virtual void | updateDensity () |
Activities, Standard States, and Activity Concentrations | |
| virtual void | getLNActivityCoefficients (doublereal *const lnac) const |
Partial Molar Properties of the Solution | |
| virtual void | getPartialMolarIntEnergies (doublereal *ubar) const |
| Return an array of partial molar internal energies for the species in the mixture. | |
Thermodynamic Values for the Species Reference States | |
| virtual void | getIntEnergy_RT_ref (doublereal *urt) const |
| Returns the vector of nondimensional internal Energies of the reference state at the current temperature of the solution and the reference pressure for each species. | |
| virtual void | setReferenceComposition (const doublereal *const x) |
| Sets the reference composition. | |
| virtual void | getReferenceComposition (doublereal *const x) const |
| Gets the reference composition. | |
Specific Properties | |
| doublereal | enthalpy_mass () const |
| Specific enthalpy. | |
| doublereal | intEnergy_mass () const |
| Specific internal energy. | |
| doublereal | entropy_mass () const |
| Specific entropy. | |
| doublereal | gibbs_mass () const |
| Specific Gibbs function. | |
| doublereal | cp_mass () const |
| Specific heat at constant pressure. | |
| doublereal | cv_mass () const |
| Specific heat at constant volume. | |
Setting the State | |
| void | setState_TPX (doublereal t, doublereal p, const doublereal *x) |
| Set the temperature (K), pressure (Pa), and mole fractions. | |
| void | setState_TPX (doublereal t, doublereal p, compositionMap &x) |
| Set the temperature (K), pressure (Pa), and mole fractions. | |
| void | setState_TPX (doublereal t, doublereal p, const std::string &x) |
| Set the temperature (K), pressure (Pa), and mole fractions. | |
| void | setState_TPY (doublereal t, doublereal p, const doublereal *y) |
| Set the internally storred temperature (K), pressure (Pa), and mass fractions of the phase. | |
| void | setState_TPY (doublereal t, doublereal p, compositionMap &y) |
| Set the internally storred temperature (K), pressure (Pa), and mass fractions of the phase. | |
| void | setState_TPY (doublereal t, doublereal p, const std::string &y) |
| Set the internally storred temperature (K), pressure (Pa), and mass fractions of the phase. | |
| void | setState_PX (doublereal p, doublereal *x) |
| Set the pressure (Pa) and mole fractions. | |
| void | setState_PY (doublereal p, doublereal *y) |
| Set the internally storred pressure (Pa) and mass fractions. | |
| virtual void | setState_HP (doublereal h, doublereal p, doublereal tol=1.e-4) |
| Set the internally storred specific enthalpy (J/kg) and pressure (Pa) of the phase. | |
| virtual void | setState_UV (doublereal u, doublereal v, doublereal tol=1.e-4) |
| Set the specific internal energy (J/kg) and specific volume (m^3/kg). | |
| virtual void | setState_SP (doublereal s, doublereal p, doublereal tol=1.e-4) |
| Set the specific entropy (J/kg/K) and pressure (Pa). | |
| virtual void | setState_SV (doublereal s, doublereal v, doublereal tol=1.e-4) |
| Set the specific entropy (J/kg/K) and specific volume (m^3/kg). | |
Chemical Equilibrium | |
| void | setElementPotentials (const vector_fp &lambda) |
| Stores the element potentials in the ThermoPhase object. | |
| bool | getElementPotentials (doublereal *lambda) const |
| Returns the element potentials storred in the ThermoPhase object. | |
Saturation Properties. | |
| virtual doublereal | satTemperature (doublereal p) const |
| Return the saturation temperature given the pressure. | |
| virtual doublereal | satPressure (doublereal t) const |
| Return the saturation pressure given the temperature. | |
| virtual doublereal | vaporFraction () const |
| Return the fraction of vapor at the current conditions. | |
| virtual void | setState_Tsat (doublereal t, doublereal x) |
| Set the state to a saturated system at a particular temperature. | |
| virtual void | setState_Psat (doublereal p, doublereal x) |
| Set the state to a saturated system at a particular pressure. | |
Initialization Methods - For Internal Use (ThermoPhase) | |
| void | saveSpeciesData (const int k, const XML_Node *const data) |
| Store a reference pointer to the XML tree containing the species data for this phase. | |
| const std::vector< const XML_Node * > & | speciesData () const |
| Return a pointer to the vector of XML nodes containing the species data for this phase. | |
| void | setSpeciesThermo (SpeciesThermo *spthermo) |
| Install a species thermodynamic property manager. | |
| virtual void | initThermoFile (std::string inputFile, std::string id) |
| int | index () const |
| void | setIndex (int m) |
Element Information | |
| std::string | elementName (int m) const |
| Name of the element with index m. | |
| int | elementIndex (std::string name) const |
| Index of element named 'name'. | |
| doublereal | atomicWeight (int m) const |
| Atomic weight of element m. | |
| doublereal | entropyElement298 (int m) const |
| Entropy of the element in its standard state at 298 K and 1 bar. | |
| int | atomicNumber (int m) const |
| Atomic number of element m. | |
| const std::vector< std::string > & | elementNames () const |
| Return a read-only reference to the vector of element names. | |
| const vector_fp & | atomicWeights () const |
| Return a read-only reference to the vector of atomic weights. | |
| int | nElements () const |
| Number of elements. | |
Adding Elements and Species | |
| void | addElement (const std::string &symbol, doublereal weight) |
| Add an element. | |
| void | addElement (const XML_Node &e) |
| Add an element from an XML specification. | |
| void | addUniqueElement (const std::string &symbol, doublereal weight, int atomicNumber=0, doublereal entropy298=ENTROPY298_UNKNOWN) |
| Add an element, checking for uniqueness. | |
| void | addUniqueElement (const XML_Node &e) |
| Adde an element, checking for uniqueness. | |
| void | addElementsFromXML (const XML_Node &phase) |
| Add all elements referenced in an XML_Node tree. | |
| void | freezeElements () |
| Prohibit addition of more elements, and prepare to add species. | |
| bool | elementsFrozen () |
| True if freezeElements has been called. | |
Adding Species | |
| void | addSpecies (const std::string &name, const doublereal *comp, doublereal charge=0.0, doublereal size=1.0) |
| void | addUniqueSpecies (const std::string &name, const doublereal *comp, doublereal charge=0.0, doublereal size=1.0) |
| Add a species to the phase, checking for uniqueness of the name. | |
| int | speciesIndex (std::string name) const |
| Index of species named 'name'. | |
| std::string | speciesName (int k) const |
| Name of the species with index k. | |
| const std::vector< std::string > & | speciesNames () const |
| Return a const referernce to the vector of species names. | |
| doublereal | size (int k) const |
| This routine returns the size of species k. | |
| bool | speciesFrozen () |
| True if freezeSpecies has been called. | |
| void | clear () |
| Remove all elements and species. | |
Composition | |
| void | getMoleFractions (doublereal *const x) const |
| Get the species mole fraction vector. | |
| virtual void | setMoleFractions (const doublereal *const x) |
| Set the mole fractions to the specified values, and then normalize them so that they sum to 1.0. | |
| virtual void | setMoleFractions_NoNorm (const doublereal *const x) |
| Set the mole fractions to the specified values without normalizing. | |
| void | getMassFractions (doublereal *const y) const |
| Get the species mass fractions. | |
| virtual void | setMassFractions (const doublereal *const y) |
| Set the mass fractions to the specified values, and then normalize them so that they sum to 1.0. | |
| virtual void | setMassFractions_NoNorm (const doublereal *const y) |
| Set the mass fractions to the specified values without normalizing. | |
| void | getConcentrations (doublereal *const c) const |
| Get the species concentrations (kmol/m^3). | |
| doublereal | concentration (const int k) const |
| Concentration of species k. | |
| virtual void | setConcentrations (const doublereal *const conc) |
| Set the concentrations to the specified values within the phase. | |
| const doublereal * | massFractions () const |
| Returns a read-only pointer to the start of the massFraction array. | |
| const doublereal * | moleFractdivMMW () const |
| Returns a read-only pointer to the start of the moleFraction/MW array. | |
Mean Properties | |
| doublereal | mean_X (const doublereal *const Q) const |
| Evaluate the mole-fraction-weighted mean of Q:
| |
| doublereal | mean_Y (const doublereal *const Q) const |
| Evaluate the mass-fraction-weighted mean of Q:
| |
| doublereal | meanMolecularWeight () const |
| The mean molecular weight. | |
| doublereal | sum_xlogx () const |
Evaluate 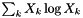 . . | |
| doublereal | sum_xlogQ (doublereal *const Q) const |
Evaluate 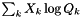 . . | |
Thermodynamic Properties | |
Class State only stores enough thermodynamic data to specify the state. In addition to composition information, it stores the temperature and mass density. | |
| doublereal | temperature () const |
| Temperature (K). | |
| virtual doublereal | density () const |
| Density (kg/m^3). | |
| doublereal | molarDensity () const |
| Molar density (kmol/m^3). | |
Public Attributes | |
| int | IMS_typeCutoff_ |
| Cutoff type. | |
| doublereal | IMS_X_o_cutoff_ |
| value of the solute mole fraction that centers the cutoff polynomials for the cutoff =1 process; | |
| doublereal | IMS_gamma_o_min_ |
| gamma_o value for the cutoff process at the zero solvent point | |
| doublereal | IMS_gamma_k_min_ |
| gamma_k minimun for the cutoff process at the zero solvent point | |
| doublereal | IMS_cCut_ |
| Parameter in the polyExp cutoff treatment having to do with rate of exp decay. | |
| doublereal | IMS_slopefCut_ |
| Parameter in the polyExp cutoff treatment. | |
| doublereal | IMS_dfCut_ |
| Parameter in the polyExp cutoff treatment having to do with rate of exp decay. | |
| doublereal | IMS_efCut_ |
| Parameter in the polyExp cutoff treatment having to do with rate of exp decay. | |
| doublereal | IMS_afCut_ |
| Parameter in the polyExp cutoff treatment having to do with rate of exp decay. | |
| doublereal | IMS_bfCut_ |
| Parameter in the polyExp cutoff treatment having to do with rate of exp decay. | |
| doublereal | IMS_slopegCut_ |
| Parameter in the polyExp cutoff treatment. | |
| doublereal | IMS_dgCut_ |
| Parameter in the polyExp cutoff treatment having to do with rate of exp decay. | |
| doublereal | IMS_egCut_ |
| Parameter in the polyExp cutoff treatment having to do with rate of exp decay. | |
| doublereal | IMS_agCut_ |
| Parameter in the polyExp cutoff treatment having to do with rate of exp decay. | |
| doublereal | IMS_bgCut_ |
| Parameter in the polyExp cutoff treatment having to do with rate of exp decay. | |
Protected Member Functions | |
| virtual void | getUnscaledMolalityActivityCoefficients (doublereal *acMolality) const |
| Get the array of unscaled non-dimensional molality based activity coefficients at the current solution temperature, pressure, and solution concentration. | |
| virtual void | applyphScale (doublereal *acMolality) const |
| Apply the current phScale to a set of activity Coefficients or activities. | |
| void | init (const array_fp &mw) |
| void | setMolecularWeight (const int k, const double mw) |
| Set the molecular weight of a single species to a given value. | |
Protected Attributes | |
| array_fp | m_speciesMolarVolume |
Species molar volume  . . | |
| int | m_formGC |
| The standard concentrations can have three different forms depending on the value of the member attribute m_formGC, which is supplied in the XML file. | |
| int | m_indexSolvent |
| Index of the solvent. | |
| int | m_pHScalingType |
| Scaling to be used for output of single-ion species activity coefficients. | |
| int | m_indexCLM |
| Index of the phScale species. | |
| doublereal | m_weightSolvent |
| Molecular weight of the Solvent. | |
| doublereal | m_xmolSolventMIN |
| doublereal | m_Mnaught |
| This is the multiplication factor that goes inside log expressions involving the molalities of species. | |
| vector_fp | m_molalities |
| Current value of the molalities of the species in the phase. | |
| doublereal | m_Pcurrent |
| Current value of the pressure - state variable. | |
| doublereal | m_Tlast_ss |
| The last temperature at which the standard statethermodynamic properties were calculated at. | |
| doublereal | m_Plast_ss |
| The last pressure at which the Standard State thermodynamic properties were calculated at. | |
| doublereal | m_P0 |
| VPSSMgr * | m_VPSS_ptr |
| Pointer to the VPSS manager that calculates all of the standard state info efficiently. | |
| std::vector< PDSS * > | m_PDSS_storage |
| Storage for the PDSS objects for the species. | |
| SpeciesThermo * | m_spthermo |
| Pointer to the calculation manager for species reference-state thermodynamic properties. | |
| std::vector< const XML_Node * > | m_speciesData |
| Vector of pointers to the species databases. | |
| int | m_index |
| Index number of the phase. | |
| doublereal | m_phi |
| Storred value of the electric potential for this phase. | |
| vector_fp | m_lambdaRRT |
| Vector of element potentials. | |
| bool | m_hasElementPotentials |
| Boolean indicating whether there is a valid set of saved element potentials for this phase. | |
| bool | m_chargeNeutralityNecessary |
| Boolean indicating whether a charge neutrality condition is a necessity. | |
| int | m_ssConvention |
| Contains the standard state convention. | |
| std::vector< doublereal > | xMol_Ref |
| Reference Mole Fraction Composition. | |
| int | m_kk |
| m_kk = Number of species in the phase. | |
| int | m_ndim |
| m_ndim is the dimensionality of the phase. | |
| vector_fp | m_weight |
| Vector of molecular weights of the species. | |
| bool | m_speciesFrozen |
| Boolean indicating whether the number of species has been frozen. | |
| Elements * | m_Elements |
| std::vector< std::string > | m_speciesNames |
| Vector of the species names. | |
| vector_fp | m_speciesComp |
| Atomic composition of the species. | |
| vector_fp | m_speciesCharge |
| m_speciesCharge: Vector of species charges length = m_kk | |
| vector_fp | m_speciesSize |
| m_speciesSize(): Vector of species sizes. | |
Private Member Functions | |
| doublereal | err (std::string msg) const |
| Internal error message. | |
| void | s_updateIMS_lnMolalityActCoeff () const |
| This function will be called to update the internally storred natural logarithm of the molality activity coefficients. | |
| void | initLengths () |
| This internal function adjusts the lengths of arrays. | |
| void | calcIMSCutoffParams_ () |
| Calculate parameters for cutoff treatments of activity coefficients. | |
Private Attributes | |
| vector_fp | m_expg0_RT |
| Vector containing the species reference exp(-G/RT) functions at T = m_tlast. | |
| vector_fp | m_pe |
| Vector of potential energies for the species. | |
| vector_fp | m_pp |
| Temporary array used in equilibrium calculations. | |
| vector_fp | m_tmpV |
| vector of size m_kk, used as a temporary holding area. | |
| vector_fp | IMS_lnActCoeffMolal_ |
| Logarithm of the molal activity coefficients. | |
Mechanical Equation of State Properties ------------------------- | |
In this equation of state implementation, the density is a function only of the mole fractions. Therefore, it can't be an independent variable. Instead, the pressure is used as the independent variable. Functions which try to set the thermodynamic state by calling setDensity() may cause an exception to be thrown. | |
| virtual void | setPressure (doublereal p) |
| Set the pressure at constant temperature. | |
| void | setDensity (const doublereal rho) |
| Overwritten setDensity() function is necessary because the density is not an indendent variable. | |
| void | setMolarDensity (const doublereal rho) |
| Overwritten setMolarDensity() function is necessary because the density is not an indendent variable. | |
| virtual void | setState_TP (doublereal t, doublereal p) |
| Set the temperature (K) and pressure (Pa). | |
| virtual doublereal | isothermalCompressibility () const |
| The isothermal compressibility. Units: 1/Pa. | |
| virtual doublereal | thermalExpansionCoeff () const |
| The thermal expansion coefficient. Units: 1/K. | |
| void | calcDensity () |
| Calculate the density of the mixture using the partial molar volumes and mole fractions as input. | |
Properties of the Standard State of the Species in the Solution | |
|
Within VPStandardStateTP, these properties are calculated via a common routine, _updateStandardStateThermo(), which must be overloaded in inherited objects. The values are cached within this object, and are not recalculated unless the temperature or pressure changes. | |
| virtual void | getStandardChemPotentials (doublereal *mu) const |
| Get the array of chemical potentials at unit activity. | |
| virtual void | getEnthalpy_RT (doublereal *hrt) const |
| Get the nondimensional Enthalpy functions for the species at their standard states at the current T and P of the solution. | |
| virtual void | getEntropy_R (doublereal *sr) const |
| Get the array of nondimensional Enthalpy functions for the standard state species at the current T and P of the solution. | |
| virtual void | getGibbs_RT (doublereal *grt) const |
| Get the nondimensional Gibbs functions for the species at their standard states of solution at the current T and P of the solution. | |
| void | getPureGibbs (doublereal *gpure) const |
| Get the nondimensional Gibbs functions for the standard state of the species at the current T and P. | |
| virtual void | getIntEnergy_RT (doublereal *urt) const |
| Returns the vector of nondimensional internal Energies of the standard state at the current temperature and pressure of the solution for each species. | |
| virtual void | getCp_R (doublereal *cpr) const |
| Get the nondimensional Heat Capacities at constant pressure for the standard state of the species at the current T and P. | |
| virtual void | getStandardVolumes (doublereal *vol) const |
| Get the molar volumes of each species in their standard states at the current T and P of the solution. | |
| virtual void | setTemperature (const doublereal temp) |
| Set the temperature of the phase. | |
| doublereal | pressure () const |
| Returns the current pressure of the phase. | |
| virtual void | updateStandardStateThermo () const |
| Updates the standard state thermodynamic functions at the current T and P of the solution. | |
| virtual void | _updateStandardStateThermo () const |
| Updates the standard state thermodynamic functions at the current T and P of the solution. | |
Thermodynamic Values for the Species Reference States (VPStandardStateTP) | |
|
| |
| virtual void | getEnthalpy_RT_ref (doublereal *hrt) const |
| Returns the vector of nondimensional enthalpies of the reference state at the current temperature of the solution and the reference pressure for the species. | |
| virtual void | getGibbs_RT_ref (doublereal *grt) const |
| Returns the vector of nondimensional Gibbs free energies of the reference state at the current temperature of the solution and the reference pressure for the species. | |
| virtual void | getGibbs_ref (doublereal *g) const |
| virtual void | getEntropy_R_ref (doublereal *er) const |
| virtual void | getCp_R_ref (doublereal *cprt) const |
| virtual void | getStandardVolumes_ref (doublereal *vol) const |
| Get the molar volumes of the species reference states at the current T and P_ref of the solution. | |
| const vector_fp & | Gibbs_RT_ref () const |
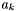 of a species in solution is related to the chemical potential by
of a species in solution is related to the chemical potential by ![\[ \mu_k = \mu_k^0(T) + \hat R T \log a_k. \]](form_98.png)
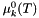 is the chemical potential at unit activity, which depends only on temperature and the pressure.
is the chemical potential at unit activity, which depends only on temperature and the pressure. 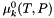 is the chemical potential at unit activity, which depends only on temperature and pressure.
is the chemical potential at unit activity, which depends only on temperature and pressure. ![\[ \mu_k = \mu_k^0(T,P) + \hat R T \log a_k. \]](form_478.png)
Detailed Description
This phase is based upon the mixing-rule assumption that all molality-based activity coefficients are equal to one.
This is a full instanteation of a ThermoPhase object. The assumption is that the molality-based activity coefficient is equal to one. This also implies that the osmotic coefficient is equal to one.
Note, this does not mean that the solution is an ideal solution. In fact, there is a singularity in the formulation as the solvent concentration goes to zero.
The mechanical equation of state is currently assumed to be that of an incompressible solution. This may change in the future. Each species has its own molar volume. The molar volume is a constant.
Class IdealMolalSoln represents a condensed phase. The phase and the pure species phases which comprise the standard states of the species are assumed to have zero volume expansivity and zero isothermal compressibility. Each species does, however, have constant but distinct partial molar volumes equal to their pure species molar volumes. The class derives from class ThermoPhase, and overloads the virtual methods defined there with ones that use expressions appropriate for incompressible mixtures.
The standard concentrations can have three different forms depending on the value of the member attribute m_formGC, which is supplied in the XML file.
| m_formGC | ActivityConc | StandardConc |
| 0 | 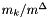 | 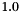 |
| 1 | 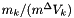 | 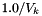 |
| 2 | 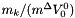 | 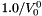 |
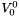 is the solvent standard molar volume.
is the solvent standard molar volume. 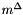 is a constant equal to a molality of
is a constant equal to a molality of 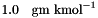 .
.
The current default is to have mformGC = 2.
The value and form of the activity concentration will affect reaction rate constants involving species in this phase.
<thermo model="IdealMolalSoln">
<standardConc model="solvent_volume" />
<solvent> H2O(l) </solvent>
<activityCoefficients model="IdealMolalSoln" >
<idealMolalSolnCutoff model="polyExp">
<gamma_O_limit> 1.0E-5 <gammaOlimit>
<gamma_k_limit> 1.0E-5 <gammaklimit>
<X_o_cutoff> 0.20 </X_o_cutoff>
<C_0_param> 0.05 </C_0_param>
<slope_f_limit> 0.6 </slopefLimit>
<slope_g_limit> 0.0 </slopegLimit>
</idealMolalSolnCutoff>
</activityCoefficients>
</thermo>
Definition at line 113 of file IdealMolalSoln.h.
Constructor & Destructor Documentation
| IdealMolalSoln | ( | ) |
Constructors.
Default constructor.
Definition at line 43 of file IdealMolalSoln.cpp.
Referenced by IdealMolalSoln::duplMyselfAsThermoPhase().
| IdealMolalSoln | ( | const IdealMolalSoln & | b | ) |
Copy Constructor.
Definition at line 70 of file IdealMolalSoln.cpp.
| IdealMolalSoln | ( | std::string | inputFile, | |
| std::string | id = "" | |||
| ) |
Constructor for phase initialization.
This constructor will initialize a phase, by reading the required information from an input file.
- Parameters:
-
inputFile Name of the Input file that contains information about the phase id id of the phase within the input file
Definition at line 116 of file IdealMolalSoln.cpp.
References IdealMolalSoln::constructPhaseFile().
| IdealMolalSoln | ( | XML_Node & | phaseRef, | |
| std::string | id = "" | |||
| ) |
Constructor for phase initialization.
This constructor will initialize a phase, by reading the required information from XML_Node tree.
- Parameters:
-
phaseRef reference for an XML_Node tree that contains the information necessary to initialize the phase. id id of the phase within the input file
Definition at line 138 of file IdealMolalSoln.cpp.
References IdealMolalSoln::constructPhaseXML().
| ~IdealMolalSoln | ( | ) | [virtual] |
Destructor.
Definition at line 167 of file IdealMolalSoln.cpp.
Member Function Documentation
| doublereal _RT | ( | ) | const [inline, inherited] |
Return the Gas Constant multiplied by the current temperature.
The units are Joules kmol-1
Definition at line 1526 of file ThermoPhase.h.
References Cantera::GasConstant, and State::temperature().
Referenced by VPStandardStateTP::getChemPotentials_RT(), IdealSolnGasVPSS::getChemPotentials_RT(), WaterSSTP::getGibbs_ref(), IdealGasPhase::getGibbs_ref(), IdealSolidSolnPhase::getGibbs_RT(), IdealMolalSoln::getPartialMolarEnthalpies(), IdealSolidSolnPhase::getPureGibbs(), IdealGasPhase::getPureGibbs(), ConstDensityThermo::getPureGibbs(), VPStandardStateTP::getStandardChemPotentials(), IdealGasPhase::getStandardChemPotentials(), and IdealGasPhase::getStandardVolumes_ref().
| void _updateStandardStateThermo | ( | ) | const [protected, virtual, inherited] |
Updates the standard state thermodynamic functions at the current T and P of the solution.
For internal use only.
If m_useTmpStandardStateStorage is true, this function must be called for every call to functions in this class.
This function is responsible for updating the following internal members, when m_useTmpStandardStateStorage is true.
- m_hss_RT;
- m_cpss_R;
- m_gss_RT;
- m_sss_R;
- m_Vss
This function doesn't check to see if the temperature or pressure has changed. It automatically assumes that it has changed. If m_useTmpStandardStateStorage is not true, this function may be required to be called by child classes to update internal member data..
Definition at line 492 of file VPStandardStateTP.cpp.
References AssertThrowMsg, VPStandardStateTP::m_Pcurrent, VPStandardStateTP::m_Plast_ss, VPStandardStateTP::m_Tlast_ss, VPStandardStateTP::m_VPSS_ptr, VPSSMgr::setState_TP(), and State::temperature().
Referenced by IdealMolalSoln::getActivities(), DebyeHuckel::getActivities(), DebyeHuckel::getMolalityActivityCoefficients(), DebyeHuckel::setState_TP(), and VPStandardStateTP::updateStandardStateThermo().
| int activityConvention | ( | ) | const [virtual, inherited] |
This method returns the activity convention.
Currently, there are two activity conventions Molar-based activities Unit activity of species at either a hypothetical pure solution of the species or at a hypothetical pure ideal solution at infinite dilution cAC_CONVENTION_MOLAR 0
- default
Molality based acvtivities (unit activity of solutes at a hypothetical 1 molal solution referenced to infinite dilution at all pressures and temperatures). cAC_CONVENTION_MOLALITY 1
We set the convention to molality here.
Reimplemented from ThermoPhase.
Definition at line 430 of file MolalityVPSSTP.cpp.
References Cantera::cAC_CONVENTION_MOLALITY.
| void addElement | ( | const XML_Node & | e | ) | [inherited] |
Add an element from an XML specification.
- Parameters:
-
e Reference to the XML_Node where the element is described.
Definition at line 138 of file Constituents.cpp.
References Elements::addElement(), and Constituents::m_Elements.
| void addElement | ( | const std::string & | symbol, | |
| doublereal | weight | |||
| ) | [inherited] |
Add an element.
- Parameters:
-
symbol Atomic symbol std::string. weight Atomic mass in amu.
Definition at line 132 of file Constituents.cpp.
References Elements::addElement(), and Constituents::m_Elements.
| void addElementsFromXML | ( | const XML_Node & | phase | ) | [inherited] |
Add all elements referenced in an XML_Node tree.
- Parameters:
-
phase Reference to the top XML_Node of a phase
Definition at line 169 of file Constituents.cpp.
References Elements::addElementsFromXML(), and Constituents::m_Elements.
| void addUniqueElement | ( | const XML_Node & | e | ) | [inherited] |
Adde an element, checking for uniqueness.
The uniqueness is checked by comparing the string symbol. If not unique, nothing is done.
- Parameters:
-
e Reference to the XML_Node where the element is described.
Definition at line 164 of file Constituents.cpp.
References Elements::addUniqueElement(), and Constituents::m_Elements.
| void addUniqueElement | ( | const std::string & | symbol, | |
| doublereal | weight, | |||
| int | atomicNumber = 0, |
|||
| doublereal | entropy298 = ENTROPY298_UNKNOWN | |||
| ) | [inherited] |
Add an element, checking for uniqueness.
The uniqueness is checked by comparing the string symbol. If not unique, nothing is done.
- Parameters:
-
symbol String symbol of the element weight Atomic weight of the element (kg kmol-1). atomicNumber Atomic number of the element (unitless) entropy298 Entropy of the element at 298 K and 1 bar in its most stable form. The default is the value ENTROPY298_UNKNOWN, which is interpreted as an unknown, and if used will cause Cantera to throw an error.
Definition at line 157 of file Constituents.cpp.
References Elements::addUniqueElement(), and Constituents::m_Elements.
| void addUniqueSpecies | ( | const std::string & | name, | |
| const doublereal * | comp, | |||
| doublereal | charge = 0.0, |
|||
| doublereal | size = 1.0 | |||
| ) | [inherited] |
Add a species to the phase, checking for uniqueness of the name.
This routine checks for uniqueness of the string name. It only adds the species if it is unique.
- Parameters:
-
name String name of the species comp Double vector containing the elemental composition of the species. charge Charge of the species. Defaults to zero. size Size of the species (meters). Defaults to 1 meter.
Definition at line 357 of file Constituents.cpp.
References Constituents::m_Elements, Constituents::m_speciesCharge, Constituents::m_speciesComp, Constituents::m_speciesNames, Constituents::m_speciesSize, and Elements::nElements().
| void applyphScale | ( | doublereal * | acMolality | ) | const [protected, virtual, inherited] |
Apply the current phScale to a set of activity Coefficients or activities.
See the Eq3/6 Manual for a thorough discussion.
- Parameters:
-
acMolality input/Output vector containing the molality based activity coefficients. length: m_kk.
Reimplemented in HMWSoln.
Definition at line 679 of file MolalityVPSSTP.cpp.
References MolalityVPSSTP::err().
Referenced by MolalityVPSSTP::getMolalityActivityCoefficients().
| int atomicNumber | ( | int | m | ) | const [inherited] |
Atomic number of element m.
- Parameters:
-
m Element index
Definition at line 117 of file Constituents.cpp.
References Elements::atomicNumber(), and Constituents::m_Elements.
Referenced by MultiPhase::addPhase().
| doublereal atomicWeight | ( | int | m | ) | const [inherited] |
Atomic weight of element m.
- Parameters:
-
m Element index
Definition at line 95 of file Constituents.cpp.
References Elements::atomicWeight(), and Constituents::m_Elements.
Referenced by WaterSSTP::initThermoXML().
| const vector_fp & atomicWeights | ( | ) | const [inherited] |
Return a read-only reference to the vector of atomic weights.
Definition at line 109 of file Constituents.cpp.
References Elements::atomicWeights(), and Constituents::m_Elements.
| void calcDensity | ( | ) | [protected, virtual] |
Calculate the density of the mixture using the partial molar volumes and mole fractions as input.
The formula for this is
![\[ \rho = \frac{\sum_k{X_k W_k}}{\sum_k{X_k V_k}} \]](form_91.png)
where 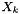 are the mole fractions,
are the mole fractions, 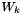 are the molecular weights, and
are the molecular weights, and 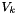 are the pure species molar volumes.
are the pure species molar volumes.
Note, the basis behind this formula is that in an ideal solution the partial molar volumes are equal to the pure species molar volumes. We have additionally specified in this class that the pure species molar volumes are independent of temperature and pressure.
NOTE: This is a non-virtual function, which is not a member of the ThermoPhase base class.
Reimplemented from VPStandardStateTP.
Definition at line 294 of file IdealMolalSoln.cpp.
References State::getMoleFractions(), IdealMolalSoln::getPartialMolarVolumes(), Phase::m_kk, IdealMolalSoln::m_pp, IdealMolalSoln::m_tmpV, State::meanMolecularWeight(), and IdealMolalSoln::setDensity().
Referenced by IdealMolalSoln::setState_TP().
| void calcIMSCutoffParams_ | ( | ) | [private] |
Calculate parameters for cutoff treatments of activity coefficients.
Some cutoff treatments for the activity coefficients actually require some calculations to create a consistent treatment.
This routine is called during the setup to calculate these parameters
Definition at line 1273 of file IdealMolalSoln.cpp.
References IdealMolalSoln::IMS_afCut_, IdealMolalSoln::IMS_agCut_, IdealMolalSoln::IMS_bfCut_, IdealMolalSoln::IMS_bgCut_, IdealMolalSoln::IMS_cCut_, IdealMolalSoln::IMS_dfCut_, IdealMolalSoln::IMS_dgCut_, IdealMolalSoln::IMS_efCut_, IdealMolalSoln::IMS_egCut_, IdealMolalSoln::IMS_gamma_k_min_, IdealMolalSoln::IMS_gamma_o_min_, IdealMolalSoln::IMS_slopefCut_, IdealMolalSoln::IMS_slopegCut_, and IdealMolalSoln::IMS_X_o_cutoff_.
Referenced by IdealMolalSoln::initThermoXML().
| void calcMolalities | ( | ) | const [inherited] |
Calculates the molality of all species and stores the result internally.
We calculate the vector of molalities of the species in the phase and store the result internally:
![\[ m_i = \frac{X_i}{1000 * M_o * X_{o,p}} \]](form_371.png)
where
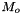 is the molecular weight of the solvent
is the molecular weight of the solvent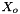 is the mole fraction of the solvent
is the mole fraction of the solvent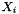 is the mole fraction of the solute.
is the mole fraction of the solute.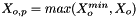
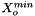 = minimum mole fraction of solvent allowed in the denominator.
= minimum mole fraction of solvent allowed in the denominator.
Definition at line 219 of file MolalityVPSSTP.cpp.
References DATA_PTR, State::getMoleFractions(), MolalityVPSSTP::m_indexSolvent, Phase::m_kk, MolalityVPSSTP::m_Mnaught, MolalityVPSSTP::m_molalities, and MolalityVPSSTP::m_xmolSolventMIN.
Referenced by DebyeHuckel::_lnactivityWaterHelgesonFixedForm(), IdealMolalSoln::getActivities(), IdealMolalSoln::getChemPotentials(), MolalityVPSSTP::getMolalities(), IdealMolalSoln::getPartialMolarEntropies(), DebyeHuckel::s_update_lnMolalityActCoeff(), IdealMolalSoln::s_updateIMS_lnMolalityActCoeff(), HMWSoln::s_updateIMS_lnMolalityActCoeff(), MolalityVPSSTP::setMolalities(), and MolalityVPSSTP::setMolalitiesByName().
| doublereal charge | ( | int | k | ) | const [inherited] |
Electrical charge of one species k molecule, divided by the magnitude of the electron charge ( 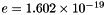 Coulombs). Dimensionless.
Coulombs). Dimensionless.
- Parameters:
-
k species index
Definition at line 266 of file Constituents.cpp.
References Constituents::m_speciesCharge.
Referenced by Phase::chargeDensity(), PDSS_HKFT::constructPDSSXML(), ThermoPhase::getElectrochemPotentials(), MolalityVPSSTP::getElectrochemPotentials(), PDSS_HKFT::initThermo(), DebyeHuckel::initThermoXML(), HMWSoln::readXMLBinarySalt(), HMWSoln::readXMLLambdaNeutral(), HMWSoln::readXMLMunnnNeutral(), HMWSoln::readXMLPsiCommonAnion(), HMWSoln::readXMLPsiCommonCation(), HMWSoln::readXMLThetaAnion(), HMWSoln::readXMLThetaCation(), HMWSoln::readXMLZetaCation(), and MolalityVPSSTP::setMolalitiesByName().
| doublereal chargeDensity | ( | ) | const [inherited] |
Charge density [C/m^3].
Definition at line 334 of file Phase.cpp.
References Constituents::charge(), Phase::moleFraction(), and Constituents::nSpecies().
| bool chargeNeutralityNecessary | ( | ) | const [inline, inherited] |
Returns the chargeNeutralityNecessity boolean.
Some phases must have zero net charge in order for their thermodynamics functions to be valid. If this is so, then the value returned from this function is true. If this is not the case, then this is false. Now, ideal gases have this parameter set to false, while solution with molality-based activity coefficients have this parameter set to true.
Definition at line 2066 of file ThermoPhase.h.
References ThermoPhase::m_chargeNeutralityNecessary.
| void clear | ( | ) | [inherited] |
Remove all elements and species.
| doublereal concentration | ( | const int | k | ) | const [inherited] |
Concentration of species k.
If k is outside the valid range, an exception will be thrown.
- Parameters:
-
k Index of species
Definition at line 134 of file State.cpp.
References State::m_dens, State::m_kk, State::m_rmolwts, and State::m_y.
| void constructPhaseFile | ( | std::string | infile, | |
| std::string | id = "" | |||
| ) |
Import and initialize an IdealMolalSoln phase specification in an XML tree into the current object.
Here we read an XML description of the phase. We import descriptions of the elements that make up the species in a phase. We import information about the species, including their reference state thermodynamic polynomials. We then freeze the state of the species.
Then, we read the species molar volumes from the xml tree to finish the initialization.
This routine is a precursor to constructPhaseXML(XML_Node*) routine, which does most of the work.
This is a virtual routine, first used here.
- Parameters:
-
infile XML file containing the description of the phase id Optional parameter identifying the name of the phase. If none is given, the first XML phase element will be used.
Definition at line 811 of file IdealMolalSoln.cpp.
References XML_Node::build(), IdealMolalSoln::constructPhaseXML(), XML_Node::copy(), Cantera::findInputFile(), Cantera::findXMLPhase(), and Phase::xml().
Referenced by IdealMolalSoln::IdealMolalSoln().
| void constructPhaseXML | ( | XML_Node & | phaseNode, | |
| std::string | id | |||
| ) |
Import and initialize an IdealMolalSoln phase specification in an XML tree into the current object.
This is the main routine for constructing the phase. It processes the XML file, and then it calls importPhase(). Then, initThermoXML() is called after importPhase().
Here we read an XML description of the phase. We import descriptions of the elements that make up the species in a phase. We import information about the species, including their reference state thermodynamic polynomials. We then freeze the state of the species.
Then, we read the species molar volumes from the xml tree to finish the initialization.
This is a virtual routine, first used in this class.
- Parameters:
-
phaseNode This object must be the phase node of a complete XML tree description of the phase, including all of the species data. In other words while "phase" must point to an XML phase object, it must have sibling nodes "speciesData" that describe the species in the phase. id ID of the phase. If nonnull, a check is done to see if phaseNode is pointing to the phase with the correct id.
Definition at line 867 of file IdealMolalSoln.cpp.
References XML_Node::hasChild(), XML_Node::id(), Cantera::importPhase(), and Constituents::size().
Referenced by IdealMolalSoln::constructPhaseFile(), and IdealMolalSoln::IdealMolalSoln().
| doublereal cp_mass | ( | ) | const [inline, inherited] |
Specific heat at constant pressure.
Units: J/kg/K.
Definition at line 1510 of file ThermoPhase.h.
References ThermoPhase::cp_mole(), and State::meanMolecularWeight().
Referenced by ThermoPhase::report(), PureFluidPhase::report(), MolalityVPSSTP::report(), SingleSpeciesTP::setState_HP(), ThermoPhase::setState_HPorUV(), SingleSpeciesTP::setState_SP(), and ThermoPhase::setState_SPorSV().
| doublereal cp_mole | ( | ) | const [virtual] |
Molar heat capacity of the solution at constant pressure. Units: J/kmol/K.
![\[ \bar{c}_p(T, P, X_k) = \sum_k X_k \bar{c}_{p,k}(T) \]](form_288.png)
Units: J/kmol/K
Reimplemented from ThermoPhase.
Definition at line 264 of file IdealMolalSoln.cpp.
References DATA_PTR, IdealMolalSoln::getPartialMolarCp(), IdealMolalSoln::m_tmpV, and State::mean_X().
| virtual doublereal critDensity | ( | ) | const [inline, virtual] |
Critical density (kg/m3).
Not implemented for this phase type.
Reimplemented from ThermoPhase.
Definition at line 763 of file IdealMolalSoln.h.
References IdealMolalSoln::err().
| virtual doublereal critPressure | ( | ) | const [inline, virtual] |
Critical pressure (Pa).
Not implemented for this phase type.
Reimplemented from ThermoPhase.
Definition at line 755 of file IdealMolalSoln.h.
References IdealMolalSoln::err().
| virtual doublereal critTemperature | ( | ) | const [inline, virtual] |
Critical temperature (K).
Not implemented for this phase type.
Reimplemented from ThermoPhase.
Definition at line 746 of file IdealMolalSoln.h.
References IdealMolalSoln::err().
| doublereal cv_mass | ( | ) | const [inline, inherited] |
Specific heat at constant volume.
Units: J/kg/K.
Definition at line 1517 of file ThermoPhase.h.
References ThermoPhase::cv_mole(), and State::meanMolecularWeight().
Referenced by ThermoPhase::report(), PureFluidPhase::report(), MolalityVPSSTP::report(), ThermoPhase::setState_HPorUV(), ThermoPhase::setState_SPorSV(), SingleSpeciesTP::setState_SV(), and SingleSpeciesTP::setState_UV().
| doublereal cv_mole | ( | ) | const [virtual] |
Molar heat capacity of the solution at constant volume. Units: J/kmol/K.
Molar heat capacity at constant volume: Units: J/kmol/K. NOT IMPLEMENTED. Units: J/kmol/K
Reimplemented from ThermoPhase.
Definition at line 275 of file IdealMolalSoln.cpp.
References IdealMolalSoln::err().
| virtual doublereal density | ( | ) | const [inline, virtual, inherited] |
Density (kg/m^3).
Reimplemented in HMWSoln.
Definition at line 314 of file State.h.
References State::m_dens.
Referenced by SingleSpeciesTP::cv_mole(), WaterSSTP::dthermalExpansionCoeffdT(), WaterSSTP::getCp_R_ref(), WaterSSTP::getEnthalpy_RT_ref(), WaterSSTP::getEntropy_R_ref(), WaterSSTP::getGibbs_RT_ref(), StoichSubstanceSSTP::getParameters(), MineralEQ3::getParameters(), MetalSHEelectrons::getParameters(), ConstDensityThermo::getParameters(), SingleSpeciesTP::getPartialMolarVolumes(), SingleSpeciesTP::getStandardVolumes(), WaterSSTP::getStandardVolumes_ref(), State::molarDensity(), ThermoPhase::report(), PureFluidPhase::report(), MolalityVPSSTP::report(), WaterSSTP::satPressure(), Phase::saveState(), IdealSolidSolnPhase::setDensity(), IdealMolalSoln::setDensity(), DebyeHuckel::setDensity(), WaterSSTP::setPressure(), WaterSSTP::setTemperature(), and WaterSSTP::vaporFraction().
| ThermoPhase * duplMyselfAsThermoPhase | ( | ) | const [virtual] |
Duplication function.
This virtual function is used to create a duplicate of the current phase. It's used to duplicate the phase when given a ThermoPhase pointer to the phase.
- Returns:
- It returns a ThermoPhase pointer.
Reimplemented from MolalityVPSSTP.
Definition at line 173 of file IdealMolalSoln.cpp.
References IdealMolalSoln::IdealMolalSoln().
| doublereal electricPotential | ( | ) | const [inline] |
Returns the electric potential of this phase (V).
Reimplemented from ThermoPhase.
Definition at line 425 of file IdealMolalSoln.h.
References ThermoPhase::m_phi.
| int elementIndex | ( | std::string | name | ) | const [inherited] |
Index of element named 'name'.
The index is an integer assigned to each element in the order it was added, beginning with 0 for the first element.
- Parameters:
-
name name of the element
If 'name' is not the name of an element in the set, then the value -1 is returned.
Definition at line 197 of file Constituents.cpp.
References Elements::elementIndex(), and Constituents::m_Elements.
Referenced by MultiPhase::init(), WaterSSTP::initThermoXML(), and PDSS_HKFT::LookupGe().
| string elementName | ( | int | m | ) | const [inherited] |
Name of the element with index m.
This is a passthrough routine to the Element object.
- Parameters:
-
m Element index.
- Exceptions:
-
If m < 0 or m >= nElements(), the exception, ElementRangeError, is thrown.
Definition at line 209 of file Constituents.cpp.
References Elements::elementName(), and Constituents::m_Elements.
Referenced by MultiPhase::addPhase(), PDSS_HKFT::convertDGFormation(), and MolalityVPSSTP::findCLMIndex().
| const vector< string > & elementNames | ( | ) | const [inherited] |
Return a read-only reference to the vector of element names.
Definition at line 229 of file Constituents.cpp.
References Elements::elementNames(), and Constituents::m_Elements.
| bool elementsFrozen | ( | ) | [inherited] |
True if freezeElements has been called.
Definition at line 183 of file Constituents.cpp.
References Elements::elementsFrozen(), and Constituents::m_Elements.
| doublereal enthalpy_mass | ( | ) | const [inline, inherited] |
Specific enthalpy.
Units: J/kg.
Definition at line 1482 of file ThermoPhase.h.
References ThermoPhase::enthalpy_mole(), and State::meanMolecularWeight().
Referenced by ThermoPhase::report(), PureFluidPhase::report(), MolalityVPSSTP::report(), SingleSpeciesTP::setState_HP(), ThermoPhase::setState_HPorUV(), and ThermoPhase::setState_SPorSV().
| doublereal enthalpy_mole | ( | ) | const [virtual] |
Molar enthalpy of the solution. Units: J/kmol.
Returns the amount of enthalpy per mole of solution. For an ideal molal solution,
![\[ \bar{h}(T, P, X_k) = \sum_k X_k \bar{h}_k(T) \]](form_281.png)
The formula is written in terms of the partial molar enthalpies.  . See the partial molar enthalpy function, getPartialMolarEnthalpies(), for details.
. See the partial molar enthalpy function, getPartialMolarEnthalpies(), for details.
Units: J/kmol
Reimplemented from ThermoPhase.
Definition at line 196 of file IdealMolalSoln.cpp.
References DATA_PTR, State::getMoleFractions(), IdealMolalSoln::getPartialMolarEnthalpies(), IdealMolalSoln::m_pp, IdealMolalSoln::m_tmpV, and State::mean_X().
| doublereal entropy_mass | ( | ) | const [inline, inherited] |
Specific entropy.
Units: J/kg/K.
Definition at line 1496 of file ThermoPhase.h.
References ThermoPhase::entropy_mole(), and State::meanMolecularWeight().
Referenced by ThermoPhase::report(), PureFluidPhase::report(), MolalityVPSSTP::report(), SingleSpeciesTP::setState_SP(), ThermoPhase::setState_SPorSV(), and SingleSpeciesTP::setState_SV().
| doublereal entropy_mole | ( | ) | const [virtual] |
Molar entropy of the solution. Units: J/kmol/K.
Returns the amount of entropy per mole of solution. For an ideal molal solution,
![\[ \bar{s}(T, P, X_k) = \sum_k X_k \bar{s}_k(T) \]](form_285.png)
The formula is written in terms of the partial molar entropies. 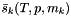 . See the partial molar entropies function, getPartialMolarEntropies(), for details.
. See the partial molar entropies function, getPartialMolarEntropies(), for details.
Units: J/kmol/K.
Reimplemented from ThermoPhase.
Definition at line 234 of file IdealMolalSoln.cpp.
References DATA_PTR, IdealMolalSoln::getPartialMolarEntropies(), IdealMolalSoln::m_tmpV, and State::mean_X().
| doublereal entropyElement298 | ( | int | m | ) | const [inherited] |
Entropy of the element in its standard state at 298 K and 1 bar.
- Parameters:
-
m Element index
Definition at line 100 of file Constituents.cpp.
References Elements::entropyElement298(), and Constituents::m_Elements.
Referenced by PDSS_HKFT::LookupGe().
| virtual int eosType | ( | ) | const [inline, virtual] |
Equation of state type flag.
The base class returns zero. Subclasses should define this to return a unique non-zero value. Constants defined for this purpose are listed in mix_defs.h.
Reimplemented from MolalityVPSSTP.
Definition at line 172 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::getUnitsStandardConc().
| doublereal err | ( | std::string | msg | ) | const [private] |
Internal error message.
- Parameters:
-
msg message to be printed
Reimplemented from MolalityVPSSTP.
Definition at line 1122 of file IdealMolalSoln.cpp.
Referenced by IdealMolalSoln::critDensity(), IdealMolalSoln::critPressure(), IdealMolalSoln::critTemperature(), IdealMolalSoln::cv_mole(), IdealMolalSoln::setPotentialEnergy(), and IdealMolalSoln::setToEquilState().
| void freezeElements | ( | ) | [inherited] |
Prohibit addition of more elements, and prepare to add species.
Definition at line 176 of file Constituents.cpp.
References Elements::freezeElements(), and Constituents::m_Elements.
| void freezeSpecies | ( | ) | [virtual, inherited] |
Finished adding species, prepare to use them for calculation of mixture properties.
Reimplemented from Constituents.
Definition at line 348 of file Phase.cpp.
References State::init(), Phase::m_data, Phase::m_kk, Constituents::molecularWeights(), and Constituents::nSpecies().
| void getActivities | ( | doublereal * | ac | ) | const [virtual] |
Get the array of non-dimensional activities at the current solution temperature, pressure, and solution concentration.
(note solvent is on molar scale)
- Parameters:
-
ac Output activity coefficients. Length: m_kk.
Reimplemented from MolalityVPSSTP.
Definition at line 503 of file IdealMolalSoln.cpp.
References VPStandardStateTP::_updateStandardStateThermo(), MolalityVPSSTP::calcMolalities(), Cantera::fmaxx(), IdealMolalSoln::IMS_lnActCoeffMolal_, IdealMolalSoln::IMS_typeCutoff_, MolalityVPSSTP::m_indexSolvent, Phase::m_kk, MolalityVPSSTP::m_molalities, MolalityVPSSTP::m_xmolSolventMIN, Phase::moleFraction(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
Referenced by IdealMolalSoln::getActivityConcentrations().
| void getActivityCoefficients | ( | doublereal * | ac | ) | const [virtual, inherited] |
Get the array of non-dimensional activity coefficients at the current solution temperature, pressure, and solution concentration.
These are mole-fraction based activity coefficients. In this object, their calculation is based on translating the values of the molality-based activity coefficients. See Denbigh p. 278 for a thorough discussion.
The molar-based activity coefficients 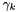 may be calculated from the molality-based activity coefficients,
may be calculated from the molality-based activity coefficients, 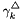 by the following formula.
by the following formula.
![\[ \gamma_k = \frac{\gamma_k^\triangle}{X_o} \]](form_366.png)
For purposes of establishing a convention, the molar activity coefficient of the solvent is set equal to the molality-based activity coefficient of the solvent:
![\[ \gamma_o = \gamma_o^\triangle \]](form_367.png)
Derived classes don't need to overload this function. This function is handled at this level.
- Parameters:
-
ac Output vector containing the mole-fraction based activity coefficients. length: m_kk.
Reimplemented from ThermoPhase.
Definition at line 465 of file MolalityVPSSTP.cpp.
References AssertThrow, MolalityVPSSTP::getMolalityActivityCoefficients(), MolalityVPSSTP::m_indexSolvent, Phase::m_kk, MolalityVPSSTP::m_xmolSolventMIN, and Phase::moleFraction().
| void getActivityConcentrations | ( | doublereal * | c | ) | const [virtual] |
This method returns an array of generalized concentrations 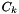 that are defined such that
that are defined such that 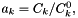 where is a standard concentration defined below. These generalized concentrations are used by kinetics manager classes to compute the forward and reverse rates of elementary reactions.
where is a standard concentration defined below. These generalized concentrations are used by kinetics manager classes to compute the forward and reverse rates of elementary reactions.
- Parameters:
-
c Array of generalized concentrations. The units depend upon the implementation of the reaction rate expressions within the phase.
Reimplemented from MolalityVPSSTP.
Definition at line 402 of file IdealMolalSoln.cpp.
References IdealMolalSoln::getActivities(), IdealMolalSoln::m_formGC, Phase::m_kk, and IdealMolalSoln::standardConcentration().
| void getAtoms | ( | int | k, | |
| double * | atomArray | |||
| ) | const [inherited] |
Get a vector containing the atomic composition of species k.
- Parameters:
-
k species index atomArray vector containing the atomic number in the species. Length: m_mm
Definition at line 480 of file Constituents.cpp.
References Constituents::m_Elements, Constituents::m_speciesComp, and Elements::nElements().
| void getChemPotentials | ( | doublereal * | mu | ) | const [virtual] |
Get the species chemical potentials: Units: J/kmol.
This function returns a vector of chemical potentials of the species in solution.
![\[ \mu_k = \mu^{o}_k(T,P) + R T \ln(\frac{m_k}{m^\Delta}) \]](form_290.png)
![\[ \mu_w = \mu^{o}_w(T,P) + R T ((X_w - 1.0) / X_w) \]](form_291.png)
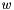 refers to the solvent species.
refers to the solvent species. 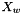 is the mole fraction of the solvent.
is the mole fraction of the solvent. 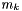 is the molality of the kth solute.
is the molality of the kth solute. 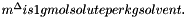
Units: J/kmol.
- Parameters:
-
mu Output vector of species chemical potentials. Length: m_kk.
Reimplemented from ThermoPhase.
Definition at line 589 of file IdealMolalSoln.cpp.
References AssertThrow, MolalityVPSSTP::calcMolalities(), Cantera::fmaxx(), Cantera::GasConstant, VPStandardStateTP::getStandardChemPotentials(), IdealMolalSoln::IMS_lnActCoeffMolal_, IdealMolalSoln::IMS_typeCutoff_, IdealMolalSoln::IMS_X_o_cutoff_, MolalityVPSSTP::m_indexSolvent, Phase::m_kk, MolalityVPSSTP::m_molalities, MAX, Phase::moleFraction(), IdealMolalSoln::s_updateIMS_lnMolalityActCoeff(), and State::temperature().
Referenced by IdealMolalSoln::gibbs_mole().
| void getChemPotentials_RT | ( | doublereal * | mu | ) | const [virtual, inherited] |
Get the array of non-dimensional species chemical potentials These are partial molar Gibbs free energies.
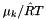 . Units: unitless
. Units: unitless
We close the loop on this function, here, calling getChemPotentials() and then dividing by RT. No need for child classes to handle.
- Parameters:
-
mu Output vector of non-dimensional species chemical potentials Length: m_kk.
Reimplemented from ThermoPhase.
Reimplemented in IdealSolnGasVPSS.
Definition at line 198 of file VPStandardStateTP.cpp.
References ThermoPhase::_RT(), ThermoPhase::getChemPotentials(), and Phase::m_kk.
| void getConcentrations | ( | doublereal *const | c | ) | const [inherited] |
Get the species concentrations (kmol/m^3).
- Parameters:
-
c On return, c contains the concentrations for all species. Array c must have a length greater than or equal to the number of species.
Definition at line 219 of file State.cpp.
References State::m_dens, State::m_ym, and Cantera::scale().
Referenced by ConstDensityThermo::getActivityCoefficients(), SurfPhase::getActivityConcentrations(), IdealSolnGasVPSS::getActivityConcentrations(), IdealGasPhase::getActivityConcentrations(), and SurfPhase::getCoverages().
| void getCp_R | ( | doublereal * | cpr | ) | const [virtual, inherited] |
Get the nondimensional Heat Capacities at constant pressure for the standard state of the species at the current T and P.
This is redefined here to call the internal function, _updateStandardStateThermo(), which calculates all standard state properties at the same time.
- Parameters:
-
cpr Output vector containing the the nondimensional Heat Capacities at constant pressure for the standard state of the species. Length: m_kk.
Reimplemented from ThermoPhase.
Definition at line 261 of file VPStandardStateTP.cpp.
References VPSSMgr::getCp_R(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
Referenced by IdealSolnGasVPSS::getPartialMolarCp(), IdealMolalSoln::getPartialMolarCp(), HMWSoln::getPartialMolarCp(), and DebyeHuckel::getPartialMolarCp().
| void getCp_R_ref | ( | doublereal * | cprt | ) | const [virtual, inherited] |
Returns the vector of nondimensional constant pressure heat capacities of the reference state at the current temperature of the solution and reference pressure for the species.
- Parameters:
-
cprt Output vector contains the nondimensional heat capacities of the species in their reference states length: m_kk, units: dimensionless.
Reimplemented from ThermoPhase.
Definition at line 330 of file VPStandardStateTP.cpp.
References VPSSMgr::getCp_R_ref(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
| virtual void getdlnActCoeffdlnC | ( | doublereal * | dlnActCoeffdlnC | ) | const [inline, virtual, inherited] |
Get the array of log concentration-like derivatives of the log activity coefficients.
This function is a virtual method. For ideal mixtures (unity activity coefficients), this can return zero. Implementations should take the derivative of the logarithm of the activity coefficient with respect to the logarithm of the concentration-like variable (i.e. mole fraction, molality, etc.) that represents the standard state. This quantity is to be used in conjunction with derivatives of that concentration-like variable when the derivative of the chemical potential is taken.
units = dimensionless
- Parameters:
-
dlnActCoeffdlnC Output vector of derivatives of the log Activity Coefficients. length = m_kk
Reimplemented from ThermoPhase.
Definition at line 141 of file VPStandardStateTP.h.
References VPStandardStateTP::err().
| void getElectrochemPotentials | ( | doublereal * | mu | ) | const [inherited] |
Get the species electrochemical potentials.
These are partial molar quantities. This method adds a term 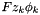 to the to each chemical potential.
to the to each chemical potential.
Units: J/kmol
- Parameters:
-
mu output vector containing the species electrochemical potentials. Length: m_kk.
Reimplemented from ThermoPhase.
Definition at line 530 of file MolalityVPSSTP.cpp.
References Constituents::charge(), ThermoPhase::electricPotential(), ThermoPhase::getChemPotentials(), and Phase::m_kk.
| bool getElementPotentials | ( | doublereal * | lambda | ) | const [inherited] |
Returns the element potentials storred in the ThermoPhase object.
Returns the storred element potentials. The element potentials are retrieved from their storred dimensionless forms by multiplying by RT.
- Parameters:
-
lambda Output vector containing the element potentials. Length = nElements. Units are Joules/kmol.
- Returns:
- bool indicating whether thare are any valid storred element potentials. The calling routine should check this bool. In the case that there aren't any, lambda is not touched.
Definition at line 1015 of file ThermoPhase.cpp.
References Cantera::GasConstant, ThermoPhase::m_hasElementPotentials, ThermoPhase::m_lambdaRRT, Constituents::nElements(), and State::temperature().
| void getEnthalpy_RT | ( | doublereal * | hrt | ) | const [inline, virtual, inherited] |
Get the nondimensional Enthalpy functions for the species at their standard states at the current T and P of the solution.
- Parameters:
-
hrt Output vector of standard state enthalpies. length = m_kk. units are unitless.
Reimplemented from ThermoPhase.
Definition at line 218 of file VPStandardStateTP.cpp.
References VPSSMgr::getEnthalpy_RT(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
Referenced by IdealSolnGasVPSS::getPartialMolarEnthalpies(), IdealMolalSoln::getPartialMolarEnthalpies(), HMWSoln::getPartialMolarEnthalpies(), and DebyeHuckel::getPartialMolarEnthalpies().
| void getEnthalpy_RT_ref | ( | doublereal * | hrt | ) | const [virtual, inherited] |
Returns the vector of nondimensional enthalpies of the reference state at the current temperature of the solution and the reference pressure for the species.
There are also temporary variables for holding the species reference-state values of Cp, H, S, and V at the last temperature and reference pressure called. These functions are not recalculated if a new call is made using the previous temperature. All calculations are done within the routine _updateRefStateThermo().
- Parameters:
-
hrt Output vector contains the nondimensional enthalpies of the reference state of the species length = m_kk, units = dimensionless.
Reimplemented from ThermoPhase.
Definition at line 280 of file VPStandardStateTP.cpp.
References VPSSMgr::getEnthalpy_RT_ref(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
| void getEntropy_R | ( | doublereal * | sr | ) | const [virtual, inherited] |
Get the array of nondimensional Enthalpy functions for the standard state species at the current T and P of the solution.
- Parameters:
-
sr Output vector of nondimensional standard state entropies. length = m_kk.
Reimplemented from ThermoPhase.
Definition at line 239 of file VPStandardStateTP.cpp.
References VPSSMgr::getEntropy_R(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
Referenced by IdealSolnGasVPSS::getPartialMolarEntropies(), IdealMolalSoln::getPartialMolarEntropies(), HMWSoln::getPartialMolarEntropies(), and DebyeHuckel::getPartialMolarEntropies().
| void getEntropy_R_ref | ( | doublereal * | er | ) | const [virtual, inherited] |
Returns the vector of nondimensional entropies of the reference state at the current temperature of the solution and the reference pressure for the species.
- Parameters:
-
er Output vector contain the nondimensional entropies of the species in their reference states length: m_kk, units: dimensionless.
Reimplemented from ThermoPhase.
Definition at line 319 of file VPStandardStateTP.cpp.
References VPSSMgr::getEntropy_R_ref(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
| void getGibbs_ref | ( | doublereal * | g | ) | const [virtual, inherited] |
Returns the vector of the gibbs function of the reference state at the current temperature of the solution and the reference pressure for the species. units = J/kmol
- Parameters:
-
g Output vector contain the Gibbs free energies of the reference state of the species length = m_kk, units = J/kmol.
Reimplemented from ThermoPhase.
Definition at line 304 of file VPStandardStateTP.cpp.
References VPSSMgr::getGibbs_ref(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
| void getGibbs_RT | ( | doublereal * | grt | ) | const [inline, virtual, inherited] |
Get the nondimensional Gibbs functions for the species at their standard states of solution at the current T and P of the solution.
- Parameters:
-
grt Output vector of nondimensional standard state Gibbs free energies. length = m_kk.
Reimplemented from ThermoPhase.
Definition at line 245 of file VPStandardStateTP.cpp.
References VPSSMgr::getGibbs_RT(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
Referenced by VPStandardStateTP::getStandardChemPotentials().
| void getGibbs_RT_ref | ( | doublereal * | grt | ) | const [virtual, inherited] |
Returns the vector of nondimensional Gibbs free energies of the reference state at the current temperature of the solution and the reference pressure for the species.
- Parameters:
-
grt Output vector contains the nondimensional Gibbs free energies of the reference state of the species length = m_kk, units = dimensionless.
Reimplemented from ThermoPhase.
Definition at line 290 of file VPStandardStateTP.cpp.
References VPSSMgr::getGibbs_RT_ref(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
| void getIntEnergy_RT | ( | doublereal * | urt | ) | const [virtual, inherited] |
Returns the vector of nondimensional internal Energies of the standard state at the current temperature and pressure of the solution for each species.
![\[ u^{ss}_k(T,P) = h^{ss}_k(T) - P * V^{ss}_k \]](form_481.png)
- Parameters:
-
urt Output vector of nondimensional standard state internal energies. length = m_kk.
Reimplemented from ThermoPhase.
Definition at line 256 of file VPStandardStateTP.cpp.
References VPSSMgr::getIntEnergy_RT(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
Referenced by IdealSolnGasVPSS::getPartialMolarIntEnergies().
| virtual void getIntEnergy_RT_ref | ( | doublereal * | urt | ) | const [inline, virtual, inherited] |
Returns the vector of nondimensional internal Energies of the reference state at the current temperature of the solution and the reference pressure for each species.
- Parameters:
-
urt Output vector of nondimensional reference state internal energies of the species. Length: m_kk
Reimplemented in IdealGasPhase, IdealSolidSolnPhase, MetalSHEelectrons, MineralEQ3, and StoichSubstanceSSTP.
Definition at line 1424 of file ThermoPhase.h.
References ThermoPhase::err().
| void getMassFractions | ( | doublereal *const | y | ) | const [inherited] |
Get the species mass fractions.
- Parameters:
-
y On return, y contains the mass fractions. Array y must have a length greater than or equal to the number of species. y Output vector of mass fractions. Length is m_kk.
Definition at line 235 of file State.cpp.
References State::m_y.
Referenced by ThermoPhase::report(), PureFluidPhase::report(), and Phase::saveState().
| void getMolalities | ( | doublereal *const | molal | ) | const [inherited] |
This function will return the molalities of the species.
We calculate the vector of molalities of the species in the phase
where
- is the molecular weight of the solvent
- is the mole fraction of the solvent
- is the mole fraction of the solute.
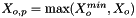
- = minimum mole fraction of solvent allowed in the denominator.
- Parameters:
-
molal Output vector of molalities. Length: m_kk.
Definition at line 246 of file MolalityVPSSTP.cpp.
References MolalityVPSSTP::calcMolalities(), Phase::m_kk, and MolalityVPSSTP::m_molalities.
Referenced by MolalityVPSSTP::report(), and vcs_MultiPhaseEquil::reportCSV().
| void getMolalityActivityCoefficients | ( | doublereal * | acMolality | ) | const [virtual] |
Get the array of non-dimensional molality-based activity coefficients at the current solution temperature, pressure, and solution concentration.
(note solvent is on molar scale. The solvent molar based activity coefficient is returned).
- Parameters:
-
acMolality Output Molality-based activity coefficients. Length: m_kk.
Reimplemented from MolalityVPSSTP.
Definition at line 546 of file IdealMolalSoln.cpp.
References Cantera::fmaxx(), IdealMolalSoln::IMS_lnActCoeffMolal_, IdealMolalSoln::IMS_typeCutoff_, MolalityVPSSTP::m_indexSolvent, Phase::m_kk, MolalityVPSSTP::m_xmolSolventMIN, Phase::moleFraction(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| void getMolecularWeights | ( | doublereal * | weights | ) | const [inherited] |
Copy the vector of molecular weights into array weights.
- Parameters:
-
weights Output array of molecular weights (kg/kmol)
Definition at line 289 of file Phase.cpp.
References Phase::molecularWeights().
| void getMolecularWeights | ( | int | iwt, | |
| doublereal * | weights | |||
| ) | const [inherited] |
Copy the vector of molecular weights into array weights.
- Parameters:
-
iwt Unused. weights Output array of molecular weights (kg/kmol)
Definition at line 281 of file Phase.cpp.
References Phase::molecularWeights().
| void getMolecularWeights | ( | vector_fp & | weights | ) | const [inherited] |
Copy the vector of molecular weights into vector weights.
- Parameters:
-
weights Output vector of molecular weights (kg/kmol)
Definition at line 271 of file Phase.cpp.
References Phase::molecularWeights().
| void getMoleFractions | ( | doublereal *const | x | ) | const [inherited] |
Get the species mole fraction vector.
- Parameters:
-
x On return, x contains the mole fractions. Must have a length greater than or equal to the number of species.
Definition at line 231 of file State.cpp.
References State::m_mmw, State::m_ym, and Cantera::scale().
Referenced by IdealMolalSoln::calcDensity(), DebyeHuckel::calcDensity(), MolalityVPSSTP::calcMolalities(), IdealMolalSoln::enthalpy_mole(), LatticePhase::getActivityCoefficients(), HMWSoln::relative_enthalpy(), ThermoPhase::report(), PureFluidPhase::report(), MolalityVPSSTP::report(), MolalityVPSSTP::setMolalitiesByName(), MultiPhase::setMoles(), ThermoPhase::setReferenceComposition(), and MultiPhase::uploadMoleFractionsFromPhases().
| void getMoleFractionsByName | ( | compositionMap & | x | ) | const [inherited] |
Get the mole fractions by name.
- Parameters:
-
x Output composition map containing the species mole fractions.
Definition at line 306 of file Phase.cpp.
References Phase::moleFraction(), Constituents::nSpecies(), and Constituents::speciesName().
| void getParameters | ( | int & | n, | |
| doublereal *const | c | |||
| ) | const [virtual] |
For internal use only.
Get the parameters used to initialize the phase.
- Parameters:
-
n number of parameters (output) c array of n coefficients
Reimplemented from ThermoPhase.
Definition at line 1090 of file IdealMolalSoln.cpp.
| void getPartialMolarCp | ( | doublereal * | cpbar | ) | const [virtual] |
Partial molar heat capacity of the solution:. UnitsL J/kmol/K.
The kth partial molar heat capacity is equal to the temperature derivative of the partial molar enthalpy of the kth species in the solution at constant P and composition (p. 220 Smith and Van Ness).
![\[ \bar{Cp}_k(T,P) = {Cp}^0_k(T) \]](form_301.png)
For this solution, this is equal to the reference state heat capacities.
Units: J/kmol/K
- Parameters:
-
cpbar Output vector of partial molar heat capacities. Length: m_kk.
Reimplemented from ThermoPhase.
Definition at line 757 of file IdealMolalSoln.cpp.
References Cantera::GasConstant, VPStandardStateTP::getCp_R(), and Phase::m_kk.
Referenced by IdealMolalSoln::cp_mole().
| void getPartialMolarEnthalpies | ( | doublereal * | hbar | ) | const [virtual] |
Returns an array of partial molar enthalpies for the species in the mixture.
Units (J/kmol) For this phase, the partial molar enthalpies are equal to the species standard state enthalpies.
![\[ \bar h_k(T,P) = \hat h^{ref}_k(T) + (P - P_{ref}) \hat V^0_k \]](form_295.png)
The reference-state pure-species enthalpies, 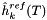 , at the reference pressure,
, at the reference pressure,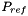 , are computed by the species thermodynamic property manager. They are polynomial functions of temperature.
, are computed by the species thermodynamic property manager. They are polynomial functions of temperature.
- See also:
- SpeciesThermo
- Parameters:
-
hbar Output vector of partial molar enthalpies. Length: m_kk.
Reimplemented from ThermoPhase.
Definition at line 655 of file IdealMolalSoln.cpp.
References ThermoPhase::_RT(), VPStandardStateTP::getEnthalpy_RT(), and Phase::m_kk.
Referenced by IdealMolalSoln::enthalpy_mole(), and IdealMolalSoln::intEnergy_mole().
| void getPartialMolarEntropies | ( | doublereal * | sbar | ) | const [virtual] |
Returns an array of partial molar entropies of the species in the solution. Units: J/kmol.
Maxwell's equations provide an insight in how to calculate this (p.215 Smith and Van Ness)
![\[ \frac{d(\mu_k)}{dT} = -\bar{s}_i \]](form_297.png)
For this phase, the partial molar entropies are equal to the standard state species entropies plus the ideal molal solution contribution.
![\[ \bar{s}_k(T,P) = s^0_k(T) - R \ln( \frac{m_k}{m^{\triangle}} ) \]](form_298.png)
![\[ \bar{s}_w(T,P) = s^0_w(T) - R ((X_w - 1.0) / X_w) \]](form_299.png)
The subscript, w, refers to the solvent species. is the mole fraction of solvent. The reference-state pure-species entropies,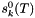 , at the reference pressure, , are computed by the species thermodynamic property manager. They are polynomial functions of temperature.
, at the reference pressure, , are computed by the species thermodynamic property manager. They are polynomial functions of temperature.
- See also:
- SpeciesThermo
- Parameters:
-
sbar Output vector of partial molar entropies. Length: m_kk.
Reimplemented from ThermoPhase.
Definition at line 690 of file IdealMolalSoln.cpp.
References MolalityVPSSTP::calcMolalities(), Cantera::fmaxx(), Cantera::GasConstant, VPStandardStateTP::getEntropy_R(), IdealMolalSoln::IMS_lnActCoeffMolal_, IdealMolalSoln::IMS_typeCutoff_, MolalityVPSSTP::m_indexSolvent, Phase::m_kk, MolalityVPSSTP::m_molalities, Phase::moleFraction(), IdealMolalSoln::s_updateIMS_lnMolalityActCoeff(), and Cantera::SmallNumber.
Referenced by IdealMolalSoln::entropy_mole().
| virtual void getPartialMolarIntEnergies | ( | doublereal * | ubar | ) | const [inline, virtual, inherited] |
Return an array of partial molar internal energies for the species in the mixture.
Units: J/kmol.
- Parameters:
-
ubar Output vector of speciar partial molar internal energies. Length = m_kk. units are J/kmol.
Reimplemented in IdealGasPhase, IdealSolnGasVPSS, and SingleSpeciesTP.
Definition at line 1244 of file ThermoPhase.h.
References ThermoPhase::err().
| void getPartialMolarVolumes | ( | doublereal * | vbar | ) | const [virtual] |
For this solution, the partial molar volumes are equal to the constant species molar volumes.
Units: m^3 kmol-1.
- Parameters:
-
vbar Output vector of partial molar volumes.
Reimplemented from ThermoPhase.
Definition at line 737 of file IdealMolalSoln.cpp.
References VPStandardStateTP::getStandardVolumes().
Referenced by IdealMolalSoln::calcDensity().
| void getPureGibbs | ( | doublereal * | gpure | ) | const [inline, virtual, inherited] |
Get the nondimensional Gibbs functions for the standard state of the species at the current T and P.
(Note resolved at this level)
- Parameters:
-
gpure Output vector of standard state Gibbs free energies. length = m_kk. units are J/kmol.
- Todo:
- This could be eliminated. It doesn't fit into the current naming convention.
Reimplemented from ThermoPhase.
Definition at line 251 of file VPStandardStateTP.cpp.
References VPSSMgr::getStandardChemPotentials(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
| void getReferenceComposition | ( | doublereal *const | x | ) | const [virtual, inherited] |
Gets the reference composition.
The reference mole fraction is a safe mole fraction.
- Parameters:
-
x Mole fraction vector containing the reference composition.
Definition at line 911 of file ThermoPhase.cpp.
References Phase::m_kk, and ThermoPhase::xMol_Ref.
| void getSpeciesMolarVolumes | ( | double * | smv | ) | const |
Fill in a return vector containing the species molar volumes units -
- Parameters:
-
smv Output vector of species molar volumes.
| void getStandardChemPotentials | ( | doublereal * | mu | ) | const [virtual, inherited] |
Get the array of chemical potentials at unit activity.
These are the standard state chemical potentials 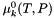 . The values are evaluated at the current temperature and pressure.
. The values are evaluated at the current temperature and pressure.
- Parameters:
-
mu Output vector of standard state chemical potentials. length = m_kk. units are J / kmol.
Reimplemented from ThermoPhase.
Definition at line 209 of file VPStandardStateTP.cpp.
References ThermoPhase::_RT(), VPStandardStateTP::getGibbs_RT(), and Phase::m_kk.
Referenced by IdealSolnGasVPSS::getChemPotentials(), IdealMolalSoln::getChemPotentials(), HMWSoln::getChemPotentials(), DebyeHuckel::getChemPotentials(), and MolalityVPSSTP::report().
| void getStandardVolumes | ( | doublereal * | vol | ) | const [virtual, inherited] |
Get the molar volumes of each species in their standard states at the current T and P of the solution.
units = m^3 / kmol
This is redefined here to call the internal function, _updateStandardStateThermo(), which calculates all standard state properties at the same time.
- Parameters:
-
vol Output vector of species volumes. length = m_kk. units = m^3 / kmol
Reimplemented from ThermoPhase.
Definition at line 266 of file VPStandardStateTP.cpp.
References VPSSMgr::getStandardVolumes(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
Referenced by IdealSolnGasVPSS::getPartialMolarVolumes(), IdealMolalSoln::getPartialMolarVolumes(), HMWSoln::getPartialMolarVolumes(), DebyeHuckel::getPartialMolarVolumes(), and HMWSoln::standardConcentration().
| void getStandardVolumes_ref | ( | doublereal * | vol | ) | const [virtual, inherited] |
Get the molar volumes of the species reference states at the current T and P_ref of the solution.
units = m^3 / kmol
- Parameters:
-
vol Output vector containing the standard state volumes. Length: m_kk.
Reimplemented from ThermoPhase.
Definition at line 341 of file VPStandardStateTP.cpp.
References VPSSMgr::getStandardVolumes_ref(), VPStandardStateTP::m_VPSS_ptr, and VPStandardStateTP::updateStandardStateThermo().
| void getUnitsStandardConc | ( | double * | uA, | |
| int | k = 0, |
|||
| int | sizeUA = 6 | |||
| ) | const [virtual] |
Returns the units of the standard and generalized concentrations Note they have the same units, as their ratio is defined to be equal to the activity of the kth species in the solution, which is unitless.
This routine is used in print out applications where the units are needed. Usually, MKS units are assumed throughout the program and in the XML input files.
- Parameters:
-
uA Output vector containing the units uA[0] = kmol units - default = 1 uA[1] = m units - default = -nDim(), the number of spatial dimensions in the Phase class. uA[2] = kg units - default = 0; uA[3] = Pa(pressure) units - default = 0; uA[4] = Temperature units - default = 0; uA[5] = time units - default = 0 k species index. Defaults to 0. sizeUA output int containing the size of the vector. Currently, this is equal to 6.
Reimplemented from MolalityVPSSTP.
Definition at line 477 of file IdealMolalSoln.cpp.
References IdealMolalSoln::eosType(), and Phase::nDim().
| void getUnscaledMolalityActivityCoefficients | ( | doublereal * | acMolality | ) | const [protected, virtual, inherited] |
Get the array of unscaled non-dimensional molality based activity coefficients at the current solution temperature, pressure, and solution concentration.
See Denbigh p. 278 for a thorough discussion. This class must be overwritten in classes which derive from MolalityVPSSTP. This function takes over from the molar-based activity coefficient calculation, getActivityCoefficients(), in derived classes.
- Parameters:
-
acMolality Output vector containing the molality based activity coefficients. length: m_kk.
Reimplemented in HMWSoln.
Definition at line 668 of file MolalityVPSSTP.cpp.
References MolalityVPSSTP::err().
Referenced by MolalityVPSSTP::getMolalityActivityCoefficients().
| doublereal gibbs_mass | ( | ) | const [inline, inherited] |
Specific Gibbs function.
Units: J/kg.
Definition at line 1503 of file ThermoPhase.h.
References ThermoPhase::gibbs_mole(), and State::meanMolecularWeight().
Referenced by ThermoPhase::report(), PureFluidPhase::report(), and MolalityVPSSTP::report().
| doublereal gibbs_mole | ( | ) | const [virtual] |
Molar Gibbs function for the solution: Units J/kmol.
Returns the gibbs free energy of the solution per mole of the solution.
![\[ \bar{g}(T, P, X_k) = \sum_k X_k \mu_k(T) \]](form_287.png)
Units: J/kmol
Reimplemented from ThermoPhase.
Definition at line 251 of file IdealMolalSoln.cpp.
References DATA_PTR, IdealMolalSoln::getChemPotentials(), IdealMolalSoln::m_tmpV, and State::mean_X().
| doublereal Hf298SS | ( | const int | k | ) | const [inline, inherited] |
Report the 298 K Heat of Formation of the standard state of one species (J kmol-1).
The 298K Heat of Formation is defined as the enthalpy change to create the standard state of the species from its constituent elements in their standard states at 298 K and 1 bar.
- Parameters:
-
k species index
- Returns:
- Returns the current value of the Heat of Formation at 298K and 1 bar
Definition at line 816 of file ThermoPhase.h.
References ThermoPhase::err().
| std::string id | ( | ) | const [inherited] |
Return the string id for the phase.
Returns the id of the phase. The ID of the phase is set to the string name of the phase within the XML file Generally, it refers to the individual model name that denotes the species, the thermo, and the reaction rate info.
Definition at line 116 of file Phase.cpp.
References Phase::m_id.
Referenced by Kinetics::kineticsSpeciesIndex(), MultiPhase::phaseIndex(), and MultiPhase::phaseName().
| int index | ( | ) | const [inline, inherited] |
For internal use only.
Index number. This method can be used to identify the location of a phase object in a list, and is used by the interface library (clib) routines for this purpose.
Reimplemented from Phase.
Definition at line 1985 of file ThermoPhase.h.
References ThermoPhase::m_index.
| void init | ( | const array_fp & | mw | ) | [protected, inherited] |
For internal use only.
Initialize. Make a local copy of the vector of molecular weights, and resize the composition arrays to the appropriate size. The only information an instance of State has about the species is their molecular weights.
- Parameters:
-
mw Vector of molecular weights of the species.
Definition at line 244 of file State.cpp.
References Cantera::int2str(), State::m_kk, State::m_mmw, State::m_molwts, State::m_rmolwts, State::m_y, State::m_ym, and Cantera::Tiny.
Referenced by Phase::freezeSpecies().
| void initLengths | ( | ) | [private] |
This internal function adjusts the lengths of arrays.
This function is not virtual nor is it inherited
Reimplemented from MolalityVPSSTP.
Definition at line 1257 of file IdealMolalSoln.cpp.
References IdealMolalSoln::IMS_lnActCoeffMolal_, IdealMolalSoln::m_expg0_RT, Phase::m_kk, IdealMolalSoln::m_pe, IdealMolalSoln::m_pp, IdealMolalSoln::m_speciesMolarVolume, IdealMolalSoln::m_tmpV, and Constituents::nSpecies().
Referenced by IdealMolalSoln::initThermo().
| void initThermo | ( | ) | [virtual] |
Initialization routine for an IdealMolalSoln phase.
This internal routine is responsible for setting up the internal storage. This is reimplemented from the ThermoPhase class.
Reimplemented from MolalityVPSSTP.
Definition at line 792 of file IdealMolalSoln.cpp.
References IdealMolalSoln::initLengths().
Referenced by IdealMolalSoln::initThermoXML().
| void initThermoFile | ( | std::string | inputFile, | |
| std::string | id | |||
| ) | [virtual, inherited] |
For internal use only.
Initialization of a ThermoPhase object using an ctml file.
This routine is a precursor to initThermoXML(XML_Node*) routine, which does most of the work. Here we read extra information about the XML description of a phase. Regular information about elements and species and their reference state thermodynamic information have already been read at this point. For example, we do not need to call this function for ideal gas equations of state.
- Parameters:
-
inputFile XML file containing the description of the phase id Optional parameter identifying the name of the phase. If none is given, the first XML phase element encountered will be used.
Definition at line 830 of file ThermoPhase.cpp.
References XML_Node::build(), XML_Node::copy(), Cantera::findInputFile(), Cantera::findXMLPhase(), ThermoPhase::initThermoXML(), and Phase::xml().
| void initThermoXML | ( | XML_Node & | phaseNode, | |
| std::string | id = "" | |||
| ) | [virtual] |
Import and initialize an IdealMolalSoln phase specification in an XML tree into the current object.
This routine is called from importPhase() to finish up the initialization of the thermo object. It reads in the species molar volumes.
- Parameters:
-
phaseNode This object must be the phase node of a complete XML tree description of the phase, including all of the species data. In other words while "phase" must point to an XML phase object, it must have sibling nodes "speciesData" that describe the species in the phase. id ID of the phase. If nonnull, a check is done to see if phaseNode is pointing to the phase with the correct id.
Reimplemented from MolalityVPSSTP.
Definition at line 915 of file IdealMolalSoln.cpp.
References XML_Node::attrib(), IdealMolalSoln::calcIMSCutoffParams_(), XML_Node::child(), XML_Node::findByAttr(), XML_Node::findByName(), Cantera::get_XML_NameID(), ctml::getFloat(), ctml::getStringArray(), XML_Node::hasChild(), XML_Node::id(), IdealMolalSoln::IMS_cCut_, IdealMolalSoln::IMS_gamma_k_min_, IdealMolalSoln::IMS_gamma_o_min_, IdealMolalSoln::IMS_slopefCut_, IdealMolalSoln::IMS_slopegCut_, IdealMolalSoln::IMS_typeCutoff_, IdealMolalSoln::IMS_X_o_cutoff_, IdealMolalSoln::initThermo(), IdealMolalSoln::m_formGC, MolalityVPSSTP::m_indexSolvent, Phase::m_kk, IdealMolalSoln::m_speciesMolarVolume, XML_Node::root(), MolalityVPSSTP::setMoleFSolventMin(), MolalityVPSSTP::setStateFromXML(), Constituents::size(), Constituents::speciesName(), and Constituents::speciesNames().
| doublereal intEnergy_mass | ( | ) | const [inline, inherited] |
Specific internal energy.
Units: J/kg.
Definition at line 1489 of file ThermoPhase.h.
References ThermoPhase::intEnergy_mole(), and State::meanMolecularWeight().
Referenced by ThermoPhase::report(), PureFluidPhase::report(), MolalityVPSSTP::report(), ThermoPhase::setState_HPorUV(), and SingleSpeciesTP::setState_UV().
| doublereal intEnergy_mole | ( | ) | const [virtual] |
Molar internal energy of the solution: Units: J/kmol.
Returns the amount of internal energy per mole of solution. For an ideal molal solution,
![\[ \bar{u}(T, P, X_k) = \sum_k X_k \bar{u}_k(T) \]](form_283.png)
The formula is written in terms of the partial molar internal energy. 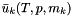 .
.
Reimplemented from ThermoPhase.
Definition at line 214 of file IdealMolalSoln.cpp.
References DATA_PTR, IdealMolalSoln::getPartialMolarEnthalpies(), IdealMolalSoln::m_tmpV, and State::mean_X().
| doublereal isothermalCompressibility | ( | ) | const [virtual] |
The isothermal compressibility. Units: 1/Pa.
The isothermal compressibility is defined as
![\[ \kappa_T = -\frac{1}{v}\left(\frac{\partial v}{\partial P}\right)_T \]](form_95.png)
It's equal to zero for this model, since the molar volume doesn't change with pressure or temperature.
Reimplemented from ThermoPhase.
Definition at line 317 of file IdealMolalSoln.cpp.
| doublereal logStandardConc | ( | int | k = 0 |
) | const [virtual] |
Returns the natural logarithm of the standard concentration of the kth species
- Parameters:
-
k Species index
Reimplemented from MolalityVPSSTP.
Definition at line 450 of file IdealMolalSoln.cpp.
References IdealMolalSoln::standardConcentration().
| doublereal massFraction | ( | std::string | name | ) | const [inherited] |
Return the mass fraction of a single species.
- Parameters:
-
name String name of the species
- Returns:
- Mass Fraction of the species
Definition at line 328 of file Phase.cpp.
References State::massFractions(), and Constituents::speciesIndex().
| doublereal massFraction | ( | int | k | ) | const [inherited] |
| const doublereal* massFractions | ( | ) | const [inline, inherited] |
Returns a read-only pointer to the start of the massFraction array.
The pointer returned is readonly
- Returns:
- returns a pointer to a vector of doubles of length m_kk.
Definition at line 242 of file State.h.
References State::m_y.
Referenced by Phase::massFraction().
| doublereal maxTemp | ( | int | k = -1 |
) | const [inline, inherited] |
Maximum temperature for which the thermodynamic data for the species are valid.
If no argument is supplied, the value returned will be the highest temperature at which the data for all species are valid. Otherwise, the value will be only for species k. This function is a wrapper that calls the species thermo maxTemp function.
- Parameters:
-
k index of the species. Default is -1, which will return the min of the max value over all species.
Definition at line 845 of file ThermoPhase.h.
References ThermoPhase::m_spthermo, and SpeciesThermo::maxTemp().
Referenced by MultiPhase::addPhase(), ThermoPhase::setState_HPorUV(), and ThermoPhase::setState_SPorSV().
| doublereal mean_X | ( | const doublereal *const | Q | ) | const [inherited] |
Evaluate the mole-fraction-weighted mean of Q:
![\[ \sum_k X_k Q_k. \]](form_451.png)
Array Q should contain pure-species molar property values.
- Parameters:
-
Q input vector of length m_kk that is to be averaged.
- Returns:
- mole-freaction-weighted mean of Q
Definition at line 223 of file State.cpp.
References State::m_mmw, and State::m_ym.
Referenced by IdealSolnGasVPSS::cp_mole(), IdealSolidSolnPhase::cp_mole(), IdealMolalSoln::cp_mole(), IdealGasPhase::cp_mole(), DebyeHuckel::cp_mole(), LatticePhase::cv_mole(), HMWSoln::cv_mole(), ConstDensityThermo::cv_mole(), SurfPhase::enthalpy_mole(), IdealSolnGasVPSS::enthalpy_mole(), IdealSolidSolnPhase::enthalpy_mole(), IdealMolalSoln::enthalpy_mole(), IdealGasPhase::enthalpy_mole(), DebyeHuckel::enthalpy_mole(), IdealSolnGasVPSS::entropy_mole(), IdealSolidSolnPhase::entropy_mole(), IdealMolalSoln::entropy_mole(), IdealGasPhase::entropy_mole(), DebyeHuckel::entropy_mole(), IdealSolidSolnPhase::gibbs_mole(), IdealMolalSoln::gibbs_mole(), HMWSoln::gibbs_mole(), DebyeHuckel::gibbs_mole(), IdealSolidSolnPhase::intEnergy_mole(), IdealMolalSoln::intEnergy_mole(), IdealGasPhase::intEnergy_mole(), and HMWSoln::relative_enthalpy().
| doublereal mean_Y | ( | const doublereal *const | Q | ) | const [inherited] |
Evaluate the mass-fraction-weighted mean of Q:
![\[ \sum_k Y_k Q_k \]](form_452.png)
.
- Parameters:
-
Q Array Q contains a vector of species property values in mass units.
- Returns:
- Return value containing the mass-fraction-weighted mean of Q.
Definition at line 227 of file State.cpp.
References Cantera::dot(), and State::m_y.
| doublereal meanMolecularWeight | ( | ) | const [inline, inherited] |
The mean molecular weight.
Units: (kg/kmol)
Definition at line 282 of file State.h.
References State::m_mmw.
Referenced by IdealSolnGasVPSS::calcDensity(), IdealMolalSoln::calcDensity(), DebyeHuckel::calcDensity(), ThermoPhase::cp_mass(), ThermoPhase::cv_mass(), ThermoPhase::enthalpy_mass(), ThermoPhase::entropy_mass(), IdealSolidSolnPhase::getActivityConcentrations(), WaterSSTP::getStandardVolumes_ref(), ThermoPhase::gibbs_mass(), ThermoPhase::intEnergy_mass(), State::molarDensity(), ThermoPhase::report(), PureFluidPhase::report(), MolalityVPSSTP::report(), State::setMolarDensity(), and IdealGasPhase::setPressure().
| doublereal minTemp | ( | int | k = -1 |
) | const [inline, inherited] |
Minimum temperature for which the thermodynamic data for the species or phase are valid.
If no argument is supplied, the value returned will be the lowest temperature at which the data for all species are valid. Otherwise, the value will be only for species k. This function is a wrapper that calls the species thermo minTemp function.
- Parameters:
-
k index of the species. Default is -1, which will return the max of the min value over all species.
Definition at line 776 of file ThermoPhase.h.
References ThermoPhase::m_spthermo, and SpeciesThermo::minTemp().
Referenced by MultiPhase::addPhase(), ThermoPhase::setState_HPorUV(), and ThermoPhase::setState_SPorSV().
| virtual void modifyOneHf298SS | ( | const int | k, | |
| const doublereal | Hf298New | |||
| ) | [inline, virtual, inherited] |
Modify the value of the 298 K Heat of Formation of one species in the phase (J kmol-1).
The 298K heat of formation is defined as the enthalpy change to create the standard state of the species from its constituent elements in their standard states at 298 K and 1 bar.
- Parameters:
-
k Species k Hf298New Specify the new value of the Heat of Formation at 298K and 1 bar
Definition at line 828 of file ThermoPhase.h.
References ThermoPhase::err().
| doublereal molarDensity | ( | ) | const [inherited] |
Molar density (kmol/m^3).
Definition at line 192 of file State.cpp.
References State::density(), and State::meanMolecularWeight().
Referenced by IdealSolidSolnPhase::enthalpy_mole(), ConstDensityThermo::getChemPotentials(), StoichSubstanceSSTP::getEnthalpy_RT(), MineralEQ3::getEnthalpy_RT(), StoichSubstanceSSTP::getIntEnergy_RT(), MineralEQ3::getIntEnergy_RT(), StoichSubstanceSSTP::getIntEnergy_RT_ref(), MineralEQ3::getIntEnergy_RT_ref(), MetalSHEelectrons::getIntEnergy_RT_ref(), IdealGasPhase::getPartialMolarVolumes(), IdealGasPhase::getStandardVolumes(), IdealSolnGasVPSS::intEnergy_mole(), IdealSolidSolnPhase::intEnergy_mole(), HMWSoln::intEnergy_mole(), DebyeHuckel::intEnergy_mole(), ConstDensityThermo::logStandardConc(), IdealGasPhase::pressure(), IdealMolalSoln::setMolarDensity(), and DebyeHuckel::setMolarDensity().
| doublereal molarMass | ( | int | k | ) | const [inline, inherited] |
Return the Molar mass of species k.
Preferred name for molecular weight.
- Parameters:
-
k index for species
- Returns:
- Return the molar mass of species k kg/kmol.
Definition at line 240 of file Constituents.h.
References Constituents::molecularWeight().
| doublereal molecularWeight | ( | int | k | ) | const [inherited] |
Molecular weight of species k.
- Parameters:
-
k index of species k
- Returns:
- Returns the molecular weight of species
k.
Definition at line 240 of file Constituents.cpp.
References Constituents::m_weight, and Constituents::nSpecies().
Referenced by VPSSMgr_Water_HKFT::_updateRefStateThermo(), VPSSMgr_Water_ConstVol::_updateRefStateThermo(), VPSSMgr_Water_HKFT::_updateStandardStateThermo(), VPSSMgr_Water_ConstVol::_updateStandardStateThermo(), SingleSpeciesTP::cv_mole(), SingleSpeciesTP::getPartialMolarVolumes(), SingleSpeciesTP::getStandardVolumes(), VPSSMgr_Water_ConstVol::getStandardVolumes_ref(), PDSS::initThermo(), VPSSMgr_Water_HKFT::initThermoXML(), VPSSMgr_Water_ConstVol::initThermoXML(), PDSS_SSVol::initThermoXML(), PDSS_ConstVol::initThermoXML(), MineralEQ3::initThermoXML(), Constituents::molarMass(), and MolalityVPSSTP::setSolvent().
| const array_fp & molecularWeights | ( | ) | const [inherited] |
Return a const reference to the internal vector of molecular weights.
Reimplemented from Constituents.
Definition at line 298 of file Phase.cpp.
Referenced by Phase::getMolecularWeights().
| const doublereal * moleFractdivMMW | ( | ) | const [inherited] |
Returns a read-only pointer to the start of the moleFraction/MW array.
This array is the array of mole fractions, each divided by the mean molecular weight.
Definition at line 215 of file State.cpp.
References State::m_ym.
Referenced by IdealSolnGasVPSS::calcDensity(), IdealSolidSolnPhase::calcDensity(), and IdealSolidSolnPhase::getActivityConcentrations().
| doublereal moleFraction | ( | std::string | name | ) | const [inherited] |
Return the mole fraction of a single species.
- Parameters:
-
name String name of the species
- Returns:
- Mole fraction of the species
Definition at line 318 of file Phase.cpp.
References Phase::moleFraction(), and Constituents::speciesIndex().
| doublereal moleFraction | ( | int | k | ) | const [inherited] |
Return the mole fraction of a single species.
- Parameters:
-
k String name of the species
- Returns:
- Mole fraction of the species
Reimplemented from State.
Definition at line 314 of file Phase.cpp.
Referenced by Phase::chargeDensity(), IdealMolalSoln::getActivities(), HMWSoln::getActivities(), DebyeHuckel::getActivities(), MolalityVPSSTP::getActivityCoefficients(), IdealSolnGasVPSS::getActivityConcentrations(), IdealSolnGasVPSS::getChemPotentials(), IdealSolidSolnPhase::getChemPotentials(), IdealMolalSoln::getChemPotentials(), IdealGasPhase::getChemPotentials(), HMWSoln::getChemPotentials(), DebyeHuckel::getChemPotentials(), IdealSolidSolnPhase::getChemPotentials_RT(), IdealMolalSoln::getMolalityActivityCoefficients(), Phase::getMoleFractionsByName(), IdealSolnGasVPSS::getPartialMolarEntropies(), IdealSolidSolnPhase::getPartialMolarEntropies(), IdealMolalSoln::getPartialMolarEntropies(), IdealGasPhase::getPartialMolarEntropies(), HMWSoln::getPartialMolarEntropies(), DebyeHuckel::getPartialMolarEntropies(), Phase::moleFraction(), DebyeHuckel::s_update_d2lnMolalityActCoeff_dT2(), DebyeHuckel::s_update_dlnMolalityActCoeff_dP(), DebyeHuckel::s_update_dlnMolalityActCoeff_dT(), DebyeHuckel::s_update_lnMolalityActCoeff(), IdealMolalSoln::s_updateIMS_lnMolalityActCoeff(), HMWSoln::s_updateIMS_lnMolalityActCoeff(), and HMWSoln::s_updatePitzer_lnMolalityActCoeff().
| doublereal moleFSolventMin | ( | ) | const [inherited] |
Returns the minimum mole fraction in the molality formulation.
Definition at line 200 of file MolalityVPSSTP.cpp.
References MolalityVPSSTP::m_xmolSolventMIN.
| std::string name | ( | ) | const [inherited] |
Return the name of the phase.
Returns the name of the phase. The name of the phase is set to the string name of the phase within the XML file Generally, it refers to the individual model name that denotes the species, the thermo, and the reaction rate info. It may also refer more specifically to a location within the domain.
Definition at line 124 of file Phase.cpp.
References Phase::m_name.
Referenced by Cantera::operator<<(), ThermoPhase::report(), PureFluidPhase::report(), MolalityVPSSTP::report(), and vcs_MultiPhaseEquil::reportCSV().
| doublereal nAtoms | ( | int | k, | |
| int | m | |||
| ) | const [inherited] |
Number of atoms of element m in species k.
- Parameters:
-
k species index m element index
Definition at line 463 of file Constituents.cpp.
References Constituents::m_Elements, Constituents::m_speciesComp, Constituents::nElements(), Elements::nElements(), and Constituents::nSpecies().
Referenced by PDSS_HKFT::convertDGFormation(), MolalityVPSSTP::findCLMIndex(), MultiPhase::init(), and IdealSolidSolnPhase::setToEquilState().
| int nDim | ( | ) | const [inline, inherited] |
Returns the number of spatial dimensions (1, 2, or 3).
Definition at line 475 of file Phase.h.
References Phase::m_ndim.
Referenced by Kinetics::addPhase(), ThermoPhase::getUnitsStandardConc(), MolalityVPSSTP::getUnitsStandardConc(), IdealSolnGasVPSS::getUnitsStandardConc(), IdealSolidSolnPhase::getUnitsStandardConc(), IdealMolalSoln::getUnitsStandardConc(), HMWSoln::getUnitsStandardConc(), and DebyeHuckel::getUnitsStandardConc().
| int nElements | ( | ) | const [inherited] |
Number of elements.
Definition at line 88 of file Constituents.cpp.
References Constituents::m_Elements, and Elements::nElements().
Referenced by MultiPhase::addPhase(), PDSS_HKFT::convertDGFormation(), MolalityVPSSTP::findCLMIndex(), ThermoPhase::getElementPotentials(), IdealSolidSolnPhase::initLengths(), IdealGasPhase::initThermo(), Constituents::nAtoms(), and ThermoPhase::setElementPotentials().
| int nSpecies | ( | ) | const [inline, inherited] |
Returns the number of species in the phase.
Definition at line 222 of file Constituents.h.
References Constituents::m_kk.
Referenced by MultiPhase::addPhase(), MultiPhase::calcElemAbundances(), Phase::chargeDensity(), vcs_MultiPhaseEquil::equilibrate_TP(), Phase::freezeSpecies(), ThermoPhase::getActivities(), Phase::getMoleFractionsByName(), MultiPhase::getMoles(), MultiPhase::init(), VPStandardStateTP::initLengths(), VPSSMgr::initLengths(), MolalityVPSSTP::initLengths(), IdealSolnGasVPSS::initLengths(), IdealSolidSolnPhase::initLengths(), IdealMolalSoln::initLengths(), HMWSoln::initLengths(), DebyeHuckel::initLengths(), StoichSubstanceSSTP::initThermo(), SingleSpeciesTP::initThermo(), Constituents::molecularWeight(), Constituents::nAtoms(), Kinetics::nTotalSpecies(), ThermoPhase::report(), PureFluidPhase::report(), MolalityVPSSTP::report(), vcs_MultiPhaseEquil::reportCSV(), Phase::restoreState(), Phase::saveState(), Kinetics::selectPhase(), SurfPhase::setCoveragesByName(), Phase::setMassFractionsByName(), MolalityVPSSTP::setMolalitiesByName(), Phase::setMoleFractionsByName(), MultiPhase::setMoles(), MultiPhase::setPhaseMoleFractions(), ThermoPhase::setState_TPX(), ThermoPhase::setState_TPY(), Constituents::speciesName(), and MultiPhase::uploadMoleFractionsFromPhases().
| IdealMolalSoln & operator= | ( | const IdealMolalSoln & | b | ) |
Assignment operator.
Definition at line 87 of file IdealMolalSoln.cpp.
References IdealMolalSoln::IMS_afCut_, IdealMolalSoln::IMS_agCut_, IdealMolalSoln::IMS_bfCut_, IdealMolalSoln::IMS_bgCut_, IdealMolalSoln::IMS_cCut_, IdealMolalSoln::IMS_dfCut_, IdealMolalSoln::IMS_dgCut_, IdealMolalSoln::IMS_efCut_, IdealMolalSoln::IMS_egCut_, IdealMolalSoln::IMS_gamma_k_min_, IdealMolalSoln::IMS_gamma_o_min_, IdealMolalSoln::IMS_lnActCoeffMolal_, IdealMolalSoln::IMS_slopefCut_, IdealMolalSoln::IMS_slopegCut_, IdealMolalSoln::IMS_typeCutoff_, IdealMolalSoln::IMS_X_o_cutoff_, IdealMolalSoln::m_expg0_RT, IdealMolalSoln::m_formGC, IdealMolalSoln::m_pe, IdealMolalSoln::m_pp, IdealMolalSoln::m_speciesMolarVolume, and IdealMolalSoln::m_tmpV.
| doublereal osmoticCoefficient | ( | ) | const [virtual, inherited] |
Calculate the osmotic coefficient.
![\[ \phi = \frac{- ln(a_o)}{\tilde{M}_o \sum_{i \ne o} m_i} \]](form_363.png)
Note there are a few of definitions of the osmotic coefficient floating around. We use the one defined in (Activity Coefficients in Electrolyte Solutions, K. S. Pitzer CRC Press, Boca Raton, 1991, p. 85, Eqn. 28). This definition is most clearly related to theoretical calculation.
units = dimensionless
Definition at line 508 of file MolalityVPSSTP.cpp.
References DATA_PTR, Cantera::fmaxx(), MolalityVPSSTP::getActivities(), MolalityVPSSTP::m_indexSolvent, Phase::m_kk, MolalityVPSSTP::m_Mnaught, and MolalityVPSSTP::m_molalities.
| int pHScale | ( | ) | const [inherited] |
Reports the pH scale, which determines the scale for single-ion activity coefficients.
Single ion activity coefficients are not unique in terms of the representing actual measureable quantities.
- Returns:
- Return the pHscale type
Definition at line 153 of file MolalityVPSSTP.cpp.
References MolalityVPSSTP::m_pHScalingType.
| doublereal pressure | ( | ) | const [inline, virtual, inherited] |
Returns the current pressure of the phase.
The pressure is an independent variable in this phase. Its current value is storred in the object VPStandardStateTP.
- Returns:
- return the pressure in pascals.
Reimplemented from ThermoPhase.
Reimplemented in DebyeHuckel, and HMWSoln.
Definition at line 345 of file VPStandardStateTP.h.
References VPStandardStateTP::m_Pcurrent.
Referenced by IdealSolnGasVPSS::intEnergy_mole(), MolalityVPSSTP::report(), and IdealSolnGasVPSS::standardConcentration().
| VPSSMgr * provideVPSSMgr | ( | ) | [inherited] |
Return a pointer to the VPSSMgr for this phase.
- Returns:
- Returns a pointer to the VPSSMgr for this phase
Definition at line 474 of file VPStandardStateTP.cpp.
References VPStandardStateTP::m_VPSS_ptr.
Referenced by PDSS::initThermo(), and PDSS::PDSS().
| bool ready | ( | ) | const [virtual, inherited] |
True if both elements and species have been frozen.
Reimplemented from Constituents.
Definition at line 363 of file Phase.cpp.
References Phase::m_kk, State::ready(), and Constituents::ready().
| doublereal refPressure | ( | ) | const [inline, inherited] |
Returns the reference pressure in Pa.
This function is a wrapper that calls the species thermo refPressure function.
Definition at line 759 of file ThermoPhase.h.
References ThermoPhase::m_spthermo, and SpeciesThermo::refPressure().
Referenced by IdealSolidSolnPhase::initLengths(), StoichSubstanceSSTP::initThermo(), SingleSpeciesTP::initThermo(), and IdealGasPhase::initThermo().
| std::string report | ( | bool | show_thermo = true |
) | const [virtual, inherited] |
returns a summary of the state of the phase as a string
Format a summary of the mixture state for output.
- Parameters:
-
show_thermo If true, extra information is printed out about the thermodynamic state of the system.
Reimplemented from ThermoPhase.
Definition at line 788 of file MolalityVPSSTP.cpp.
References ThermoPhase::cp_mass(), ThermoPhase::cp_mole(), ThermoPhase::cv_mass(), ThermoPhase::cv_mole(), State::density(), ThermoPhase::electricPotential(), ThermoPhase::enthalpy_mass(), ThermoPhase::enthalpy_mole(), ThermoPhase::entropy_mass(), ThermoPhase::entropy_mole(), MolalityVPSSTP::getActivities(), ThermoPhase::getChemPotentials(), MolalityVPSSTP::getMolalities(), MolalityVPSSTP::getMolalityActivityCoefficients(), State::getMoleFractions(), VPStandardStateTP::getStandardChemPotentials(), ThermoPhase::gibbs_mass(), ThermoPhase::gibbs_mole(), ThermoPhase::intEnergy_mass(), ThermoPhase::intEnergy_mole(), State::meanMolecularWeight(), Phase::name(), Constituents::nSpecies(), VPStandardStateTP::pressure(), Cantera::SmallNumber, Constituents::speciesIndex(), Constituents::speciesName(), and State::temperature().
| void restoreState | ( | int | lenstate, | |
| const doublereal * | state | |||
| ) | [inherited] |
Restore the state of the phase from a previously saved state vector.
- Parameters:
-
lenstate Length of the state vector state Vector of state conditions.
Definition at line 154 of file Phase.cpp.
References Constituents::nSpecies(), State::setDensity(), State::setMassFractions_NoNorm(), and State::setTemperature().
| void restoreState | ( | const vector_fp & | state | ) | [inherited] |
| void s_updateIMS_lnMolalityActCoeff | ( | ) | const [private] |
This function will be called to update the internally storred natural logarithm of the molality activity coefficients.
Normally the solutes are all zero. However, sometimes they are not, due to stability schemes
Definition at line 1140 of file IdealMolalSoln.cpp.
References MolalityVPSSTP::calcMolalities(), IdealMolalSoln::IMS_afCut_, IdealMolalSoln::IMS_agCut_, IdealMolalSoln::IMS_bfCut_, IdealMolalSoln::IMS_bgCut_, IdealMolalSoln::IMS_cCut_, IdealMolalSoln::IMS_dfCut_, IdealMolalSoln::IMS_dgCut_, IdealMolalSoln::IMS_efCut_, IdealMolalSoln::IMS_egCut_, IdealMolalSoln::IMS_gamma_k_min_, IdealMolalSoln::IMS_gamma_o_min_, IdealMolalSoln::IMS_lnActCoeffMolal_, IdealMolalSoln::IMS_typeCutoff_, IdealMolalSoln::IMS_X_o_cutoff_, MolalityVPSSTP::m_indexSolvent, Phase::m_kk, MolalityVPSSTP::m_xmolSolventMIN, MAX, and Phase::moleFraction().
Referenced by IdealMolalSoln::getActivities(), IdealMolalSoln::getChemPotentials(), IdealMolalSoln::getMolalityActivityCoefficients(), and IdealMolalSoln::getPartialMolarEntropies().
| virtual doublereal satPressure | ( | doublereal | t | ) | const [inline, virtual, inherited] |
Return the saturation pressure given the temperature.
- Parameters:
-
t Temperature (Kelvin)
Reimplemented in DebyeHuckel, HMWSoln, PureFluidPhase, SingleSpeciesTP, and WaterSSTP.
Definition at line 1817 of file ThermoPhase.h.
References ThermoPhase::err().
| virtual doublereal satTemperature | ( | doublereal | p | ) | const [inline, virtual, inherited] |
Return the saturation temperature given the pressure.
- Parameters:
-
p Pressure (Pa)
Reimplemented in DebyeHuckel, HMWSoln, PureFluidPhase, and SingleSpeciesTP.
Definition at line 1809 of file ThermoPhase.h.
References ThermoPhase::err().
| void saveSpeciesData | ( | const int | k, | |
| const XML_Node *const | data | |||
| ) | [inherited] |
Store a reference pointer to the XML tree containing the species data for this phase.
The following methods are used in the process of constructing the phase and setting its parameters from a specification in an input file. They are not normally used in application programs. To see how they are used, see files importCTML.cpp and ThermoFactory.cpp.
This is used to access data needed to construct transport manager later.
For internal use only.
- Parameters:
-
k Species index data Pointer to the XML_Node data containing information about the species in the phase.
Definition at line 941 of file ThermoPhase.cpp.
References ThermoPhase::m_speciesData.
| void saveState | ( | int | lenstate, | |
| doublereal * | state | |||
| ) | const [inherited] |
Write to array 'state' the current internal state.
- Parameters:
-
lenstate length of the state array. Must be >= nSpecies() + 2 state output vector. Must be of length nSpecies() + 2 or greater.
Definition at line 144 of file Phase.cpp.
References State::density(), State::getMassFractions(), and State::temperature().
| void saveState | ( | vector_fp & | state | ) | const [inherited] |
Save the current internal state of the phase.
Write to vector 'state' the current internal state.
- Parameters:
-
state output vector. Will be resized to nSpecies() + 2 on return.
Definition at line 140 of file Phase.cpp.
References Constituents::nSpecies().
| void setConcentrations | ( | const doublereal *const | conc | ) | [virtual, inherited] |
Set the concentrations to the specified values within the phase.
We set the concentrations here and therefore we set the overall density of the phase. We hold the temperature constant during this operation. Therefore, we have possibly changed the pressure of the phase by calling this routine.
- Parameters:
-
conc The input vector to this routine is in dimensional units. For volumetric phases c[k] is the concentration of the kth species in kmol/m3. For surface phases, c[k] is the concentration in kmol/m2. The length of the vector is the number of species in the phase.
Reimplemented in IdealSolidSolnPhase.
Definition at line 196 of file State.cpp.
References State::m_kk, State::m_mmw, State::m_molwts, State::m_y, State::m_ym, State::setDensity(), and State::stateMFChangeCalc().
Referenced by SurfPhase::setCoverages(), and SurfPhase::setCoveragesNoNorm().
| void setDensity | ( | const doublereal | rho | ) | [virtual] |
Overwritten setDensity() function is necessary because the density is not an indendent variable.
This function will now throw an error condition
For internal use only.
May have to adjust the strategy here to make the eos for these materials slightly compressible, in order to create a condition where the density is a function of the pressure.
This function will now throw an error condition.
NOTE: This is an overwritten function from the State.h class
- Parameters:
-
rho Input Density
Reimplemented from State.
Definition at line 352 of file IdealMolalSoln.cpp.
References State::density().
Referenced by IdealMolalSoln::calcDensity().
| void setElectricPotential | ( | doublereal | v | ) | [inline] |
Set the electric potential of this phase (V).
This is used by classes InterfaceKinetics and EdgeKinetics to compute the rates of charge-transfer reactions, and in computing the electrochemical potentials of the species.
Each phase may have its own electric potential.
- Parameters:
-
v Input value of the electric potential in Volts
Reimplemented from ThermoPhase.
Definition at line 420 of file IdealMolalSoln.h.
References ThermoPhase::m_phi.
| void setElementPotentials | ( | const vector_fp & | lambda | ) | [inherited] |
Stores the element potentials in the ThermoPhase object.
Called by function 'equilibrate' in ChemEquil.h to transfer the element potentials to this object after every successful equilibration routine. The element potentials are storred in their dimensionless forms, calculated by dividing by RT.
- Parameters:
-
lambda Input vector containing the element potentials. Length = nElements. Units are Joules/kmol.
Definition at line 993 of file ThermoPhase.cpp.
References Cantera::GasConstant, ThermoPhase::m_hasElementPotentials, ThermoPhase::m_lambdaRRT, Constituents::nElements(), and State::temperature().
| void setID | ( | std::string | id | ) | [inherited] |
Set the string id for the phase.
Sets the id of the phase. The ID of the phase is originally set to the string name of the phase within the XML file. Generally, it refers to the individual model name that denotes the species, the thermo, and the reaction rate info.
- Parameters:
-
id String id of the phase
Definition at line 120 of file Phase.cpp.
References Phase::m_id.
| void setIndex | ( | int | m | ) | [inline, inherited] |
For internal use only.
Set the index number. The Cantera interface library uses this method to set the index number to the location of the pointer to this object in the pointer array it maintains. Using this method for any other purpose will lead to unpredictable results if used in conjunction with the interface library.
- Parameters:
-
m Input the index number.
Reimplemented from Phase.
Definition at line 1998 of file ThermoPhase.h.
References ThermoPhase::m_index.
| void setMassFractions | ( | const doublereal *const | y | ) | [virtual, inherited] |
Set the mass fractions to the specified values, and then normalize them so that they sum to 1.0.
- Parameters:
-
y Array of unnormalized mass fraction values (input). Must have a length greater than or equal to the number of species. y Input vector of mass fractions. There is no restriction on the sum of the mass fraction vector. Internally, the State object will normalize this vector before storring its contents. Length is m_kk.
Reimplemented in IdealSolidSolnPhase.
Definition at line 142 of file State.cpp.
References State::m_kk, State::m_mmw, State::m_rmolwts, State::m_y, State::m_ym, Cantera::scale(), and State::stateMFChangeCalc().
Referenced by Phase::setMassFractionsByName(), ThermoPhase::setState_PY(), Phase::setState_RY(), ThermoPhase::setState_TPY(), Phase::setState_TRY(), and Phase::setState_TY().
| void setMassFractions_NoNorm | ( | const doublereal *const | y | ) | [virtual, inherited] |
Set the mass fractions to the specified values without normalizing.
This is useful when the normalization condition is being handled by some other means, for example by a constraint equation as part of a larger set of equations.
- Parameters:
-
y Input vector of mass fractions. Length is m_kk.
Reimplemented in IdealSolidSolnPhase.
Definition at line 167 of file State.cpp.
References State::m_kk, State::m_mmw, State::m_rmolwts, State::m_y, State::m_ym, and State::stateMFChangeCalc().
Referenced by Phase::restoreState().
| void setMassFractionsByName | ( | const std::string & | x | ) | [inherited] |
Set the species mass fractions by name.
Species not listed by name in x are set to zero.
- Parameters:
-
x String containing a composition map
Definition at line 204 of file Phase.cpp.
References Constituents::nSpecies(), Cantera::parseCompString(), Phase::setMassFractionsByName(), and Constituents::speciesName().
| void setMassFractionsByName | ( | compositionMap & | yMap | ) | [inherited] |
Set the species mass fractions by name.
- Parameters:
-
yMap map from species names to mass fraction values. Species not listed by name in yMapare set to zero.
Definition at line 193 of file Phase.cpp.
References Constituents::nSpecies(), State::setMassFractions(), and Constituents::speciesName().
Referenced by Phase::setMassFractionsByName(), ThermoPhase::setState_TPY(), Phase::setState_TRY(), and ThermoPhase::setStateFromXML().
| void setMolalities | ( | const doublereal *const | molal | ) | [inherited] |
Set the molalities of the solutes in a phase.
Note, the entry for the solvent is not used. We are supplied with the molalities of all of the solute species. We then calculate the mole fractions of all species and update the ThermoPhase object.
![\[ m_i = \frac{X_i}{M_o/1000 * X_{o,p}} \]](form_376.png)
where
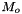 is the molecular weight of the solvent
is the molecular weight of the solvent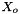 is the mole fraction of the solvent
is the mole fraction of the solvent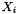 is the mole fraction of the solute.
is the mole fraction of the solute.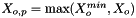
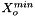 = minimum mole fraction of solvent allowed in the denominator.
= minimum mole fraction of solvent allowed in the denominator.
The formulas for calculating mole fractions are
![\[ L^{sum} = \frac{1}{\tilde{M}_o X_o} = \frac{1}{\tilde{M}_o} + \sum_{i\ne o} m_i \]](form_359.png)
Then,
![\[ X_o = \frac{1}{\tilde{M}_o L^{sum}} \]](form_360.png)
![\[ X_i = \frac{m_i}{L^{sum}} \]](form_361.png)
It is currently an error if the solvent mole fraction is attempted to be set to a value lower than .
- Parameters:
-
molal Input vector of molalities. Length: m_kk.
Definition at line 268 of file MolalityVPSSTP.cpp.
References MolalityVPSSTP::calcMolalities(), DATA_PTR, MolalityVPSSTP::m_indexSolvent, Phase::m_kk, MolalityVPSSTP::m_Mnaught, MolalityVPSSTP::m_molalities, and State::setMoleFractions().
Referenced by MolalityVPSSTP::setState_TPM().
| void setMolalitiesByName | ( | const std::string & | name | ) | [inherited] |
Set the molalities of a phase.
Set the molalities of the solutes in a phase. Note, the entry for the solvent is not used.
- Parameters:
-
name String containing the information for a composition map.
Definition at line 391 of file MolalityVPSSTP.cpp.
References Constituents::nSpecies(), Cantera::parseCompString(), MolalityVPSSTP::setMolalitiesByName(), and Constituents::speciesName().
| void setMolalitiesByName | ( | compositionMap & | xMap | ) | [inherited] |
Set the molalities of a phase.
Set the molalities of the solutes in a phase. Note, the entry for the solvent is not used.
- Parameters:
-
xMap Composition Map containing the molalities.
Definition at line 305 of file MolalityVPSSTP.cpp.
References MolalityVPSSTP::calcMolalities(), Constituents::charge(), DATA_PTR, State::getMoleFractions(), MolalityVPSSTP::m_indexSolvent, MolalityVPSSTP::m_Mnaught, MolalityVPSSTP::m_xmolSolventMIN, Cantera::max(), Constituents::nSpecies(), State::setMoleFractions(), and Constituents::speciesName().
Referenced by MolalityVPSSTP::setMolalitiesByName(), MolalityVPSSTP::setState_TPM(), and MolalityVPSSTP::setStateFromXML().
| void setMolarDensity | ( | const doublereal | rho | ) | [virtual] |
Overwritten setMolarDensity() function is necessary because the density is not an indendent variable.
This function will now throw an error condition.
NOTE: This is an overwritten function from the State.h class
- Parameters:
-
rho Input Density
Reimplemented from State.
Definition at line 369 of file IdealMolalSoln.cpp.
References State::molarDensity().
| void setMolecularWeight | ( | const int | k, | |
| const double | mw | |||
| ) | [inline, protected, inherited] |
Set the molecular weight of a single species to a given value.
- Parameters:
-
k id of the species mw Molecular Weight (kg kmol-1)
Definition at line 385 of file State.h.
References State::m_molwts, and State::m_rmolwts.
Referenced by WaterSSTP::initThermoXML().
| void setMoleFractions | ( | const doublereal *const | x | ) | [virtual, inherited] |
Set the mole fractions to the specified values, and then normalize them so that they sum to 1.0.
- Parameters:
-
x Array of unnormalized mole fraction values (input). Must have a length greater than or equal to the number of species. x Input vector of mole fractions. There is no restriction on the sum of the mole fraction vector. Internally, the State object will normalize this vector before storring its contents. Length is m_kk.
Reimplemented in IdealSolidSolnPhase.
Definition at line 102 of file State.cpp.
References Cantera::dot(), State::m_kk, State::m_mmw, State::m_molwts, State::m_y, State::m_ym, and State::stateMFChangeCalc().
Referenced by SingleSpeciesTP::initThermo(), WaterSSTP::initThermoXML(), MolalityVPSSTP::setMolalities(), MolalityVPSSTP::setMolalitiesByName(), Phase::setMoleFractionsByName(), ThermoPhase::setState_PX(), Phase::setState_RX(), Phase::setState_TNX(), ThermoPhase::setState_TPX(), Phase::setState_TRX(), and Phase::setState_TX().
| void setMoleFractions_NoNorm | ( | const doublereal *const | x | ) | [virtual, inherited] |
Set the mole fractions to the specified values without normalizing.
This is useful when the normalization condition is being handled by some other means, for example by a constraint equation as part of a larger set of equations.
- Parameters:
-
x Input vector of mole fractions. Length is m_kk.
Reimplemented in IdealSolidSolnPhase.
Definition at line 115 of file State.cpp.
References Cantera::dot(), State::m_kk, State::m_mmw, State::m_molwts, State::m_y, State::m_ym, and State::stateMFChangeCalc().
| void setMoleFractionsByName | ( | const std::string & | x | ) | [inherited] |
Set the mole fractions of a group of species by name.
The string x is in the form of a composition map Species which are not listed by name in the composition map are set to zero.
- Parameters:
-
x string x in the form of a composition map
Definition at line 177 of file Phase.cpp.
References Constituents::nSpecies(), Cantera::parseCompString(), Phase::setMoleFractionsByName(), and Constituents::speciesName().
| void setMoleFractionsByName | ( | compositionMap & | xMap | ) | [inherited] |
Set the species mole fractions by name.
- Parameters:
-
xMap map from species names to mole fraction values. Species not listed by name in xMapare set to zero.
Definition at line 166 of file Phase.cpp.
References Constituents::nSpecies(), State::setMoleFractions(), and Constituents::speciesName().
Referenced by Phase::setMoleFractionsByName(), ThermoPhase::setState_TPX(), Phase::setState_TRX(), and ThermoPhase::setStateFromXML().
| void setMoleFSolventMin | ( | doublereal | xmolSolventMIN | ) | [inherited] |
Sets the minimum mole fraction in the molality formulation.
Note the molality formulation is singular in the limit that the solvent mole fraction goes to zero. Numerically, how this limit is treated and resolved is an ongoing issue within Cantera.
- Parameters:
-
xmolSolventMIN Input double containing the minimum mole fraction
Definition at line 188 of file MolalityVPSSTP.cpp.
References MolalityVPSSTP::m_xmolSolventMIN.
Referenced by IdealMolalSoln::initThermoXML(), and HMWSoln::initThermoXML().
| void setName | ( | std::string | nm | ) | [inherited] |
Sets the string name for the phase.
Sets the name of the phase. The name of the phase is originally set to the string name of the phase within the XML file. Generally, it refers to the individual model name that denotes the species, the thermo, and the reaction rate info. It may also refer more specifically to a location within the domain.
- Parameters:
-
nm String name of the phase
Definition at line 128 of file Phase.cpp.
References Phase::m_name.
| void setNDim | ( | int | ndim | ) | [inline, inherited] |
Set the number of spatial dimensions (1, 2, or 3).
The number of spatial dimensions is used for vector involving directions.
- Parameters:
-
ndim Input number of dimensions.
Definition at line 484 of file Phase.h.
References Phase::m_ndim.
Referenced by EdgePhase::EdgePhase(), EdgePhase::operator=(), and SurfPhase::SurfPhase().
| void setParameters | ( | int | n, | |
| doublereal *const | c | |||
| ) | [virtual] |
For internal use only.
Set equation of state parameters. The number and meaning of these depends on the subclass.
- Parameters:
-
n number of parameters c array of n coefficients
Reimplemented from ThermoPhase.
Definition at line 1087 of file IdealMolalSoln.cpp.
| void setParametersFromXML | ( | const XML_Node & | eosdata | ) | [virtual] |
Set equation of state parameter values from XML entries. This method is called by function importPhase in file importCTML.cpp when processing a phase definition in an input file. It should be overloaded in subclasses to set any parameters that are specific to that particular phase model.
- Parameters:
-
eosdata An XML_Node object corresponding to the "thermo" entry for this phase in the input file.
Reimplemented from VPStandardStateTP.
Definition at line 1107 of file IdealMolalSoln.cpp.
| void setpHScale | ( | const int | pHscaleType | ) | [inherited] |
Set the pH scale, which determines the scale for single-ion activity coefficients.
Single ion activity coefficients are not unique in terms of the representing actual measureable quantities.
- Parameters:
-
pHscaleType Integer representing the pHscale
Definition at line 139 of file MolalityVPSSTP.cpp.
References Cantera::int2str(), MolalityVPSSTP::m_pHScalingType, Cantera::PHSCALE_NBS, and Cantera::PHSCALE_PITZER.
| virtual void setPotentialEnergy | ( | int | k, | |
| doublereal | pe | |||
| ) | [inline, virtual] |
Set the potential energy of species k to pe.
Units: J/kmol. This function must be reimplemented in inherited classes of ThermoPhase.
- Parameters:
-
k Species index pe Input potential energy.
Definition at line 396 of file IdealMolalSoln.h.
References IdealMolalSoln::err().
| void setPressure | ( | doublereal | p | ) | [virtual] |
Set the pressure at constant temperature.
Units: Pa. This method sets a constant within the object. The mass density is not a function of pressure.
- Parameters:
-
p Input Pressure
Reimplemented from VPStandardStateTP.
Definition at line 290 of file IdealMolalSoln.cpp.
References IdealMolalSoln::setState_TP(), and State::temperature().
| void setReferenceComposition | ( | const doublereal *const | x | ) | [virtual, inherited] |
Sets the reference composition.
- Parameters:
-
x Mole fraction vector to set the reference composition to. If this is zero, then the reference mole fraction is set to the current mole fraction vector.
Definition at line 891 of file ThermoPhase.cpp.
References DATA_PTR, State::getMoleFractions(), Phase::m_kk, and ThermoPhase::xMol_Ref.
Referenced by ThermoPhase::initThermoXML().
| void setSolvent | ( | int | k | ) | [inherited] |
This routine sets the index number of the solvent for the phase.
Note, having a solvent is a precursor to many things having to do with molality.
- Parameters:
-
k the solvent index number
Definition at line 164 of file MolalityVPSSTP.cpp.
References AssertThrowMsg, MolalityVPSSTP::m_indexSolvent, Phase::m_kk, MolalityVPSSTP::m_Mnaught, MolalityVPSSTP::m_weightSolvent, and Constituents::molecularWeight().
Referenced by MolalityVPSSTP::initThermo(), MolalityVPSSTP::initThermoXML(), and HMWSoln::initThermoXML().
| void setSpeciesThermo | ( | SpeciesThermo * | spthermo | ) | [inline, inherited] |
Install a species thermodynamic property manager.
The species thermodynamic property manager computes properties of the pure species for use in constructing solution properties. It is meant for internal use, and some classes derived from ThermoPhase may not use any species thermodynamic property manager. This method is called by function importPhase() in importCTML.cpp.
- Parameters:
-
spthermo input pointer to the species thermodynamic property manager.
For internal use only.
Definition at line 1887 of file ThermoPhase.h.
References ThermoPhase::m_spthermo.
Referenced by VPSSMgrFactory::newVPSSMgr().
| void setState_HP | ( | doublereal | h, | |
| doublereal | p, | |||
| doublereal | tol = 1.e-4 | |||
| ) | [virtual, inherited] |
Set the internally storred specific enthalpy (J/kg) and pressure (Pa) of the phase.
- Parameters:
-
h Specific enthalpy (J/kg) p Pressure (Pa) tol Optional parameter setting the tolerance of the calculation. Defaults to 1.0E-4
Reimplemented in PureFluidPhase, and SingleSpeciesTP.
Definition at line 238 of file ThermoPhase.cpp.
References ThermoPhase::setState_HPorUV().
| virtual void setState_Psat | ( | doublereal | p, | |
| doublereal | x | |||
| ) | [inline, virtual, inherited] |
Set the state to a saturated system at a particular pressure.
- Parameters:
-
p Pressure (Pa) x Fraction of vapor
Reimplemented in DebyeHuckel, HMWSoln, PureFluidPhase, and SingleSpeciesTP.
Definition at line 1840 of file ThermoPhase.h.
References ThermoPhase::err().
| void setState_PX | ( | doublereal | p, | |
| doublereal * | x | |||
| ) | [inherited] |
Set the pressure (Pa) and mole fractions.
Note, the mole fractions are set first before the pressure is set. Setting the pressure may involve the solution of a nonlinear equation.
- Parameters:
-
p Pressure (Pa) x Vector of mole fractions. Length is equal to m_kk.
Reimplemented in SingleSpeciesTP.
Definition at line 230 of file ThermoPhase.cpp.
References State::setMoleFractions(), and ThermoPhase::setPressure().
Referenced by IdealSolnGasVPSS::setToEquilState(), IdealSolidSolnPhase::setToEquilState(), and IdealGasPhase::setToEquilState().
| void setState_PY | ( | doublereal | p, | |
| doublereal * | y | |||
| ) | [inherited] |
Set the internally storred pressure (Pa) and mass fractions.
Note, the temperature is held constant during this operation. Note, the mass fractions are set first before the pressure is set. Setting the pressure may involve the solution of a nonlinear equation.
- Parameters:
-
p Pressure (Pa) y Vector of mass fractions. Length is equal to m_kk.
Reimplemented in SingleSpeciesTP.
Definition at line 234 of file ThermoPhase.cpp.
References State::setMassFractions(), and ThermoPhase::setPressure().
| void setState_RX | ( | doublereal | rho, | |
| doublereal * | x | |||
| ) | [inherited] |
Set the density (kg/m^3) and mole fractions.
- Parameters:
-
rho Density (kg/m^3) x vector of species mole fractions. Length is equal to m_kk
Definition at line 259 of file Phase.cpp.
References State::setDensity(), and State::setMoleFractions().
| void setState_RY | ( | doublereal | rho, | |
| doublereal * | y | |||
| ) | [inherited] |
Set the density (kg/m^3) and mass fractions.
- Parameters:
-
rho Density (kg/m^3) y vector of species mass fractions. Length is equal to m_kk
Definition at line 264 of file Phase.cpp.
References State::setDensity(), and State::setMassFractions().
| void setState_SP | ( | doublereal | s, | |
| doublereal | p, | |||
| doublereal | tol = 1.e-4 | |||
| ) | [virtual, inherited] |
Set the specific entropy (J/kg/K) and pressure (Pa).
This function fixes the internal state of the phase so that the specific entropy and the pressure have the value of the input parameters.
- Parameters:
-
s specific entropy (J/kg/K) p specific pressure (Pa). tol Optional parameter setting the tolerance of the calculation. Defaults to 1.0E-4
Reimplemented in PureFluidPhase, and SingleSpeciesTP.
Definition at line 512 of file ThermoPhase.cpp.
References ThermoPhase::setState_SPorSV().
| void setState_SV | ( | doublereal | s, | |
| doublereal | v, | |||
| doublereal | tol = 1.e-4 | |||
| ) | [virtual, inherited] |
Set the specific entropy (J/kg/K) and specific volume (m^3/kg).
This function fixes the internal state of the phase so that the specific entropy and specific volume have the value of the input parameters.
- Parameters:
-
s specific entropy (J/kg/K) v specific volume (m^3/kg). tol Optional parameter setting the tolerance of the calculation. Defaults to 1.0E-4
Reimplemented in PureFluidPhase, and SingleSpeciesTP.
Definition at line 517 of file ThermoPhase.cpp.
References ThermoPhase::setState_SPorSV().
| void setState_TNX | ( | doublereal | t, | |
| doublereal | n, | |||
| const doublereal * | x | |||
| ) | [inherited] |
Set the internally storred temperature (K), molar density (kmol/m^3), and mole fractions.
Note, the mole fractions are always set first, before the molar density
- Parameters:
-
t Temperature in kelvin n molar density (kmol/m^3) x vector of species mole fractions. Length is equal to m_kk
Definition at line 220 of file Phase.cpp.
References State::setMolarDensity(), State::setMoleFractions(), and State::setTemperature().
| void setState_TP | ( | doublereal | t, | |
| doublereal | p | |||
| ) | [virtual] |
Set the temperature (K) and pressure (Pa).
Set the temperature and pressure.
- Parameters:
-
t Temperature (K) p Pressure (Pa)
Reimplemented from VPStandardStateTP.
Definition at line 377 of file IdealMolalSoln.cpp.
References IdealMolalSoln::calcDensity(), VPStandardStateTP::m_Pcurrent, VPStandardStateTP::setTemperature(), and VPStandardStateTP::updateStandardStateThermo().
Referenced by IdealMolalSoln::setPressure().
| void setState_TPM | ( | doublereal | t, | |
| doublereal | p, | |||
| const std::string & | m | |||
| ) | [inherited] |
Set the temperature (K), pressure (Pa), and molalities.
- Parameters:
-
t Temperature (K) p Pressure (Pa) m String which gets translated into a composition map for the molalities of the solutes.
Definition at line 623 of file MolalityVPSSTP.cpp.
References MolalityVPSSTP::setMolalitiesByName(), and VPStandardStateTP::setState_TP().
| void setState_TPM | ( | doublereal | t, | |
| doublereal | p, | |||
| compositionMap & | m | |||
| ) | [inherited] |
Set the temperature (K), pressure (Pa), and molalities.
- Parameters:
-
t Temperature (K) p Pressure (Pa) m compositionMap containing the molalities
Definition at line 615 of file MolalityVPSSTP.cpp.
References MolalityVPSSTP::setMolalitiesByName(), and VPStandardStateTP::setState_TP().
| void setState_TPM | ( | doublereal | t, | |
| doublereal | p, | |||
| const doublereal *const | molalities | |||
| ) | [inherited] |
Set the temperature (K), pressure (Pa), and molalities (gmol kg-1) of the solutes.
- Parameters:
-
t Temperature (K) p Pressure (Pa) molalities Input vector of molalities of the solutes. Length: m_kk.
Definition at line 606 of file MolalityVPSSTP.cpp.
References MolalityVPSSTP::setMolalities(), and VPStandardStateTP::setState_TP().
| void setState_TPX | ( | doublereal | t, | |
| doublereal | p, | |||
| const std::string & | x | |||
| ) | [inherited] |
Set the temperature (K), pressure (Pa), and mole fractions.
Note, the mole fractions are set first before the pressure is set. Setting the pressure may involve the solution of a nonlinear equation.
- Parameters:
-
t Temperature (K) p Pressure (Pa) x String containing a composition map of the mole fractions. Species not in the composition map are assumed to have zero mole fraction
Reimplemented in SingleSpeciesTP.
Definition at line 186 of file ThermoPhase.cpp.
References Constituents::nSpecies(), Cantera::parseCompString(), Phase::setMoleFractionsByName(), ThermoPhase::setPressure(), State::setTemperature(), and Constituents::speciesName().
| void setState_TPX | ( | doublereal | t, | |
| doublereal | p, | |||
| compositionMap & | x | |||
| ) | [inherited] |
Set the temperature (K), pressure (Pa), and mole fractions.
Note, the mole fractions are set first before the pressure is set. Setting the pressure may involve the solution of a nonlinear equation.
- Parameters:
-
t Temperature (K) p Pressure (Pa) x Composition map of mole fractions. Species not in the composition map are assumed to have zero mole fraction
Reimplemented in SingleSpeciesTP.
Definition at line 181 of file ThermoPhase.cpp.
References Phase::setMoleFractionsByName(), ThermoPhase::setPressure(), and State::setTemperature().
| void setState_TPX | ( | doublereal | t, | |
| doublereal | p, | |||
| const doublereal * | x | |||
| ) | [inherited] |
Set the temperature (K), pressure (Pa), and mole fractions.
Note, the mole fractions are set first before the pressure is set. Setting the pressure may involve the solution of a nonlinear equation.
- Parameters:
-
t Temperature (K) p Pressure (Pa) x Vector of mole fractions. Length is equal to m_kk.
Reimplemented in SingleSpeciesTP.
Definition at line 176 of file ThermoPhase.cpp.
References State::setMoleFractions(), ThermoPhase::setPressure(), and State::setTemperature().
Referenced by MultiPhase::setMoles(), and MultiPhase::setPhaseMoleFractions().
| void setState_TPY | ( | doublereal | t, | |
| doublereal | p, | |||
| const std::string & | y | |||
| ) | [inherited] |
Set the internally storred temperature (K), pressure (Pa), and mass fractions of the phase.
Note, the mass fractions are set first before the pressure is set. Setting the pressure may involve the solution of a nonlinear equation.
- Parameters:
-
t Temperature (K) p Pressure (Pa) y String containing a composition map of the mass fractions. Species not in the composition map are assumed to have zero mass fraction
Reimplemented in SingleSpeciesTP.
Definition at line 211 of file ThermoPhase.cpp.
References Constituents::nSpecies(), Cantera::parseCompString(), Phase::setMassFractionsByName(), ThermoPhase::setPressure(), State::setTemperature(), and Constituents::speciesName().
| void setState_TPY | ( | doublereal | t, | |
| doublereal | p, | |||
| compositionMap & | y | |||
| ) | [inherited] |
Set the internally storred temperature (K), pressure (Pa), and mass fractions of the phase.
Note, the mass fractions are set first before the pressure is set. Setting the pressure may involve the solution of a nonlinear equation.
- Parameters:
-
t Temperature (K) p Pressure (Pa) y Composition map of mass fractions. Species not in the composition map are assumed to have zero mass fraction
Reimplemented in SingleSpeciesTP.
Definition at line 206 of file ThermoPhase.cpp.
References Phase::setMassFractionsByName(), ThermoPhase::setPressure(), and State::setTemperature().
| void setState_TPY | ( | doublereal | t, | |
| doublereal | p, | |||
| const doublereal * | y | |||
| ) | [inherited] |
Set the internally storred temperature (K), pressure (Pa), and mass fractions of the phase.
Note, the mass fractions are set first before the pressure is set. Setting the pressure may involve the solution of a nonlinear equation.
- Parameters:
-
t Temperature (K) p Pressure (Pa) y Vector of mass fractions. Length is equal to m_kk.
Reimplemented in SingleSpeciesTP.
Definition at line 201 of file ThermoPhase.cpp.
References State::setMassFractions(), ThermoPhase::setPressure(), and State::setTemperature().
| void setState_TR | ( | doublereal | t, | |
| doublereal | rho | |||
| ) | [inherited] |
Set the internally storred temperature (K) and density (kg/m^3).
Set the temperature (K) and density (kg/m^3).
- Parameters:
-
t Temperature in kelvin rho Density (kg/m^3)
Definition at line 244 of file Phase.cpp.
References State::setDensity(), and State::setTemperature().
| void setState_TRX | ( | doublereal | t, | |
| doublereal | dens, | |||
| compositionMap & | x | |||
| ) | [inherited] |
Set the internally storred temperature (K), density, and mole fractions.
Set the temperature (K), density (kg/m^3), and mole fractions.
Note, the mole fractions are always set first, before the density
- Parameters:
-
t Temperature in kelvin dens Density (kg/m^3) x Composition Map containing the mole fractions. Species not included in the map are assumed to have a zero mole fraction.
Definition at line 226 of file Phase.cpp.
References State::setDensity(), Phase::setMoleFractionsByName(), and State::setTemperature().
| void setState_TRX | ( | doublereal | t, | |
| doublereal | dens, | |||
| const doublereal * | x | |||
| ) | [inherited] |
Set the internally storred temperature (K), density, and mole fractions.
Set the temperature (K), density (kg/m^3), and mole fractions.
Note, the mole fractions are always set first, before the density
- Parameters:
-
t Temperature in kelvin dens Density (kg/m^3) x vector of species mole fractions. Length is equal to m_kk
Definition at line 215 of file Phase.cpp.
References State::setDensity(), State::setMoleFractions(), and State::setTemperature().
| void setState_TRY | ( | doublereal | t, | |
| doublereal | dens, | |||
| compositionMap & | y | |||
| ) | [inherited] |
Set the internally storred temperature (K), density, and mass fractions.
Set the temperature (K), density (kg/m^3), and mass fractions.
Note, the mass fractions are always set first, before the density
- Parameters:
-
t Temperature in kelvin dens Density (kg/m^3) y Composition Map containing the mass fractions. Species not included in the map are assumed to have a zero mass fraction.
Definition at line 238 of file Phase.cpp.
References State::setDensity(), Phase::setMassFractionsByName(), and State::setTemperature().
| void setState_TRY | ( | doublereal | t, | |
| doublereal | dens, | |||
| const doublereal * | y | |||
| ) | [inherited] |
Set the internally storred temperature (K), density, and mass fractions.
Set the temperature (K), density (kg/m^3), and mass fractions.
Note, the mass fractions are always set first, before the density
- Parameters:
-
t Temperature in kelvin dens Density (kg/m^3) y vector of species mass fractions. Length is equal to m_kk
Definition at line 232 of file Phase.cpp.
References State::setDensity(), State::setMassFractions(), and State::setTemperature().
| virtual void setState_Tsat | ( | doublereal | t, | |
| doublereal | x | |||
| ) | [inline, virtual, inherited] |
Set the state to a saturated system at a particular temperature.
- Parameters:
-
t Temperature (kelvin) x Fraction of vapor
Reimplemented in DebyeHuckel, HMWSoln, PureFluidPhase, and SingleSpeciesTP.
Definition at line 1831 of file ThermoPhase.h.
References ThermoPhase::err().
| void setState_TX | ( | doublereal | t, | |
| doublereal * | x | |||
| ) | [inherited] |
Set the internally storred temperature (K) and mole fractions.
Set the temperature (K) and mole fractions.
- Parameters:
-
t Temperature in kelvin x vector of species mole fractions. Length is equal to m_kk
Definition at line 249 of file Phase.cpp.
References State::setMoleFractions(), and State::setTemperature().
| void setState_TY | ( | doublereal | t, | |
| doublereal * | y | |||
| ) | [inherited] |
Set the internally storred temperature (K) and mass fractions.
Set the temperature (K) and mass fractions.
- Parameters:
-
t Temperature in kelvin y vector of species mass fractions. Length is equal to m_kk
Definition at line 254 of file Phase.cpp.
References State::setMassFractions(), and State::setTemperature().
| void setState_UV | ( | doublereal | u, | |
| doublereal | v, | |||
| doublereal | tol = 1.e-4 | |||
| ) | [virtual, inherited] |
Set the specific internal energy (J/kg) and specific volume (m^3/kg).
This function fixes the internal state of the phase so that the specific internal energy and specific volume have the value of the input parameters.
- Parameters:
-
u specific internal energy (J/kg) v specific volume (m^3/kg). tol Optional parameter setting the tolerance of the calculation. Defaults to 1.0E-4
Reimplemented in PureFluidPhase, and SingleSpeciesTP.
Definition at line 243 of file ThermoPhase.cpp.
References ThermoPhase::setState_HPorUV().
| void setStateFromXML | ( | const XML_Node & | state | ) | [virtual, inherited] |
Set equation of state parameter values from XML entries.
This method is called by function importPhase() in file importCTML.cpp when processing a phase definition in an input file. It should be overloaded in subclasses to set any parameters that are specific to that particular phase model.
The MolalityVPSSTP object defines a new method for setting the concentrations of a phase. The new method is defined by a block called "soluteMolalities". If this block is found, the concentrations within that phase are set to the "name":"molalities pairs found within that XML block. The solvent concentration is then set to everything else.
The function first calls the overloaded function , VPStandardStateTP::setStateFromXML(), to pick up the parent class behavior.
usage: Overloaded functions should call this function before carrying out their own behavior.
- Parameters:
-
state An XML_Node object corresponding to the "state" entry for this phase in the input file.
Reimplemented from ThermoPhase.
Definition at line 590 of file MolalityVPSSTP.cpp.
References ctml::getChildValue(), ctml::getFloat(), XML_Node::hasChild(), MolalityVPSSTP::setMolalitiesByName(), and VPStandardStateTP::setPressure().
Referenced by IdealMolalSoln::initThermoXML(), and DebyeHuckel::initThermoXML().
| void setTemperature | ( | const doublereal | temp | ) | [virtual, inherited] |
Set the temperature of the phase.
Currently this passes down to setState_TP(). It does not make sense to calculate the standard state without first setting T and P.
- Parameters:
-
temp Temperature (kelvin)
Reimplemented from State.
Reimplemented in DebyeHuckel, and HMWSoln.
Definition at line 376 of file VPStandardStateTP.cpp.
References VPStandardStateTP::m_Pcurrent, VPStandardStateTP::setState_TP(), and VPStandardStateTP::updateStandardStateThermo().
Referenced by VPStandardStateTP::setState_TP(), and IdealMolalSoln::setState_TP().
| virtual void setToEquilState | ( | const doublereal * | lambda_RT | ) | [inline, virtual] |
This method is used by the ChemEquil equilibrium solver.
It sets the state such that the chemical potentials satisfy
![\[ \frac{\mu_k}{\hat R T} = \sum_m A_{k,m} \left(\frac{\lambda_m} {\hat R T}\right) \]](form_18.png)
where 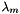 is the element potential of element m. The temperature is unchanged. Any phase (ideal or not) that implements this method can be equilibrated by ChemEquil.
is the element potential of element m. The temperature is unchanged. Any phase (ideal or not) that implements this method can be equilibrated by ChemEquil.
Not implemented.
- Parameters:
-
lambda_RT vector of Nondimensional element potentials.
Reimplemented from MolalityVPSSTP.
Definition at line 698 of file IdealMolalSoln.h.
References IdealMolalSoln::err().
| void setVPSSMgr | ( | VPSSMgr * | vp_ptr | ) | [inherited] |
set the VPSS Mgr
- Parameters:
-
vp_ptr Pointer to the manager
Definition at line 362 of file VPStandardStateTP.cpp.
References VPStandardStateTP::m_VPSS_ptr.
| doublereal size | ( | int | k | ) | const [inline, inherited] |
This routine returns the size of species k.
- Parameters:
-
k index of the species
- Returns:
- Returns the size of the species. Units are meters.
Definition at line 309 of file Constituents.h.
References Constituents::m_speciesSize.
Referenced by IdealSolidSolnPhase::constructPhaseXML(), IdealMolalSoln::constructPhaseXML(), HMWSoln::constructPhaseXML(), DebyeHuckel::constructPhaseXML(), SurfPhase::getCoverages(), SurfPhase::initThermo(), IdealMolalSoln::initThermoXML(), SurfPhase::setCoverages(), SurfPhase::setCoveragesNoNorm(), and SurfPhase::standardConcentration().
| int solventIndex | ( | ) | const [inherited] |
Returns the solvent index.
Definition at line 179 of file MolalityVPSSTP.cpp.
References MolalityVPSSTP::m_indexSolvent.
| const std::vector< const XML_Node * > & speciesData | ( | ) | const [inherited] |
Return a pointer to the vector of XML nodes containing the species data for this phase.
Return a pointer to the XML tree containing the species data for this phase.
Definition at line 950 of file ThermoPhase.cpp.
References Phase::m_kk, and ThermoPhase::m_speciesData.
Referenced by MineralEQ3::initThermoXML(), HMWSoln::initThermoXML(), and DebyeHuckel::initThermoXML().
| bool speciesFrozen | ( | ) | [inline, inherited] |
True if freezeSpecies has been called.
Definition at line 318 of file Constituents.h.
References Constituents::m_speciesFrozen.
| int speciesIndex | ( | std::string | name | ) | const [inherited] |
Index of species named 'name'.
The first species added will have index 0, and the last one index nSpecies() - 1.
- Parameters:
-
name String name of the species
- Returns:
- Returns the index of the species.
Definition at line 414 of file Constituents.cpp.
References Constituents::m_speciesNames.
Referenced by HMWSoln::HMWSoln(), HMWSoln::initThermoXML(), DebyeHuckel::initThermoXML(), Kinetics::kineticsSpeciesIndex(), Phase::massFraction(), Phase::moleFraction(), HMWSoln::readXMLBinarySalt(), HMWSoln::readXMLLambdaNeutral(), HMWSoln::readXMLMunnnNeutral(), HMWSoln::readXMLPsiCommonAnion(), HMWSoln::readXMLPsiCommonCation(), HMWSoln::readXMLThetaAnion(), HMWSoln::readXMLThetaCation(), HMWSoln::readXMLZetaCation(), MolalityVPSSTP::report(), and Kinetics::speciesPhase().
| double speciesMolarVolume | ( | int | k | ) | const |
Report the molar volume of species k.
units -
- Parameters:
-
k Species index.
| string speciesName | ( | int | k | ) | const [inherited] |
Name of the species with index k.
- Parameters:
-
k index of the species
Definition at line 434 of file Constituents.cpp.
References Constituents::m_speciesNames, and Constituents::nSpecies().
Referenced by MolalityVPSSTP::findCLMIndex(), Phase::getMoleFractionsByName(), MultiPhase::init(), IdealMolalSoln::initThermoXML(), HMWSoln::initThermoXML(), DebyeHuckel::initThermoXML(), Kinetics::kineticsSpeciesName(), HMWSoln::readXMLBinarySalt(), ThermoPhase::report(), PureFluidPhase::report(), MolalityVPSSTP::report(), vcs_MultiPhaseEquil::reportCSV(), HMWSoln::s_updatePitzer_d2lnMolalityActCoeff_dT2(), HMWSoln::s_updatePitzer_dlnMolalityActCoeff_dP(), HMWSoln::s_updatePitzer_dlnMolalityActCoeff_dT(), HMWSoln::s_updatePitzer_lnMolalityActCoeff(), SurfPhase::setCoveragesByName(), Phase::setMassFractionsByName(), MolalityVPSSTP::setMolalitiesByName(), Phase::setMoleFractionsByName(), ThermoPhase::setState_TPX(), and ThermoPhase::setState_TPY().
| const vector< string > & speciesNames | ( | ) | const [inherited] |
Return a const referernce to the vector of species names.
Definition at line 447 of file Constituents.cpp.
References Constituents::m_speciesNames.
Referenced by PDSS_SSVol::constructPDSSFile(), PDSS_HKFT::constructPDSSFile(), PDSS_ConstVol::constructPDSSFile(), VPSSMgr_Water_HKFT::initThermoXML(), VPSSMgr_Water_ConstVol::initThermoXML(), VPSSMgr_ConstVol::initThermoXML(), IdealSolidSolnPhase::initThermoXML(), IdealMolalSoln::initThermoXML(), HMWSoln::initThermoXML(), and DebyeHuckel::initThermoXML().
| SpeciesThermo& speciesThermo | ( | ) | [inline] |
Return a reference to the species thermodynamic property manager.
- Todo:
- This method will fail if no species thermo manager has been installed.
Reimplemented from ThermoPhase.
Definition at line 786 of file IdealMolalSoln.h.
References ThermoPhase::m_spthermo.
| doublereal standardConcentration | ( | int | k = 0 |
) | const [virtual] |
The standard concentration used to normalize the generalized concentration.
In many cases, this quantity will be the same for all species in a phase - for example, for an ideal gas 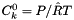 . For this reason, this method returns a single value, instead of an array. However, for phases in which the standard concentration is species-specific (e.g. surface species of different sizes), this method may be called with an optional parameter indicating the species.
. For this reason, this method returns a single value, instead of an array. However, for phases in which the standard concentration is species-specific (e.g. surface species of different sizes), this method may be called with an optional parameter indicating the species.
- Parameters:
-
k Species index
Reimplemented from MolalityVPSSTP.
Definition at line 430 of file IdealMolalSoln.cpp.
References IdealMolalSoln::m_formGC, MolalityVPSSTP::m_indexSolvent, and IdealMolalSoln::m_speciesMolarVolume.
Referenced by IdealMolalSoln::getActivityConcentrations(), and IdealMolalSoln::logStandardConc().
| int standardStateConvention | ( | ) | const [virtual, inherited] |
This method returns the convention used in specification of the standard state, of which there are currently two, temperature based, and variable pressure based.
Currently, there are two standard state conventions:
- Temperature-based activities cSS_CONVENTION_TEMPERATURE 0
- default
- Variable Pressure and Temperature -based activities cSS_CONVENTION_VPSS 1
Reimplemented from ThermoPhase.
Definition at line 169 of file VPStandardStateTP.cpp.
References Cantera::cSS_CONVENTION_VPSS.
| void stateMFChangeCalc | ( | bool | forceChange = false |
) | [inline, inherited] |
Every time the mole fractions have changed, this routine will increment the stateMFNumber.
- Parameters:
-
forceChange If this is true then the stateMFNumber always changes. This defaults to false.
Definition at line 31 of file State.cpp.
References State::m_stateNum.
Referenced by State::setConcentrations(), State::setMassFractions(), State::setMassFractions_NoNorm(), State::setMoleFractions(), and State::setMoleFractions_NoNorm().
| int stateMFNumber | ( | ) | const [inline, inherited] |
Return the state number.
Return the State Mole Fraction Number.
Definition at line 445 of file State.h.
References State::m_stateNum.
| doublereal sum_xlogQ | ( | doublereal *const | Q | ) | const [inherited] |
Evaluate .
- Parameters:
-
Q Vector of length m_kk to take the log average of
- Returns:
- Returns the indicated sum.
Definition at line 188 of file State.cpp.
References State::m_mmw, and State::m_ym.
| doublereal sum_xlogx | ( | ) | const [inherited] |
Evaluate .
- Returns:
- returns the indicated sum. units are dimensionless.
Definition at line 184 of file State.cpp.
References State::m_mmw, and State::m_ym.
Referenced by IdealSolnGasVPSS::entropy_mole(), IdealSolidSolnPhase::entropy_mole(), IdealGasPhase::entropy_mole(), and IdealSolidSolnPhase::gibbs_mole().
| doublereal temperature | ( | ) | const [inline, inherited] |
Temperature (K).
Definition at line 309 of file State.h.
References State::m_temp.
Referenced by ThermoPhase::_RT(), VPStandardStateTP::_updateStandardStateThermo(), SurfPhase::_updateThermo(), SingleSpeciesTP::_updateThermo(), IdealSolidSolnPhase::_updateThermo(), IdealGasPhase::_updateThermo(), HMWSoln::A_Debye_TP(), DebyeHuckel::A_Debye_TP(), MultiPhase::addPhase(), HMWSoln::ADebye_J(), HMWSoln::ADebye_L(), HMWSoln::ADebye_V(), IdealSolnGasVPSS::calcDensity(), HMWSoln::calcDensity(), LatticePhase::cp_mole(), ConstDensityThermo::cp_mole(), SingleSpeciesTP::cv_mole(), HMWSoln::d2A_DebyedT2_TP(), DebyeHuckel::d2A_DebyedT2_TP(), HMWSoln::dA_DebyedP_TP(), DebyeHuckel::dA_DebyedP_TP(), HMWSoln::dA_DebyedT_TP(), DebyeHuckel::dA_DebyedT_TP(), WaterSSTP::dthermalExpansionCoeffdT(), IdealSolnGasVPSS::enthalpy_mole(), IdealSolidSolnPhase::enthalpy_mole(), IdealGasPhase::enthalpy_mole(), SurfPhase::getChemPotentials(), IdealSolnGasVPSS::getChemPotentials(), IdealSolidSolnPhase::getChemPotentials(), IdealMolalSoln::getChemPotentials(), IdealGasPhase::getChemPotentials(), HMWSoln::getChemPotentials(), DebyeHuckel::getChemPotentials(), SingleSpeciesTP::getChemPotentials_RT(), IdealSolidSolnPhase::getChemPotentials_RT(), WaterSSTP::getCp_R_ref(), ThermoPhase::getElementPotentials(), WaterSSTP::getEnthalpy_RT(), SurfPhase::getEnthalpy_RT(), StoichSubstanceSSTP::getEnthalpy_RT(), MineralEQ3::getEnthalpy_RT(), IdealSolidSolnPhase::getEnthalpy_RT(), WaterSSTP::getEnthalpy_RT_ref(), WaterSSTP::getEntropy_R_ref(), SingleSpeciesTP::getGibbs_ref(), IdealSolidSolnPhase::getGibbs_ref(), WaterSSTP::getGibbs_RT(), SurfPhase::getGibbs_RT(), WaterSSTP::getGibbs_RT_ref(), StoichSubstanceSSTP::getIntEnergy_RT(), MineralEQ3::getIntEnergy_RT(), IdealSolidSolnPhase::getIntEnergy_RT(), StoichSubstanceSSTP::getIntEnergy_RT_ref(), MineralEQ3::getIntEnergy_RT_ref(), MetalSHEelectrons::getIntEnergy_RT_ref(), IdealSolidSolnPhase::getIntEnergy_RT_ref(), HMWSoln::getPartialMolarCp(), DebyeHuckel::getPartialMolarCp(), SurfPhase::getPartialMolarEnthalpies(), SingleSpeciesTP::getPartialMolarEnthalpies(), IdealSolnGasVPSS::getPartialMolarEnthalpies(), IdealSolidSolnPhase::getPartialMolarEnthalpies(), IdealGasPhase::getPartialMolarEnthalpies(), HMWSoln::getPartialMolarEnthalpies(), DebyeHuckel::getPartialMolarEnthalpies(), HMWSoln::getPartialMolarEntropies(), DebyeHuckel::getPartialMolarEntropies(), SingleSpeciesTP::getPartialMolarIntEnergies(), IdealSolnGasVPSS::getPartialMolarIntEnergies(), IdealGasPhase::getPartialMolarIntEnergies(), HMWSoln::getPartialMolarVolumes(), DebyeHuckel::getPartialMolarVolumes(), SingleSpeciesTP::getPureGibbs(), WaterSSTP::getStandardChemPotentials(), StoichSubstanceSSTP::getStandardChemPotentials(), MineralEQ3::getStandardChemPotentials(), MetalSHEelectrons::getStandardChemPotentials(), IdealGasPhase::getStandardChemPotentials(), WaterSSTP::getStandardVolumes_ref(), IdealSolnGasVPSS::gibbs_mole(), IdealSolidSolnPhase::gibbs_mole(), IdealGasPhase::gibbs_mole(), IdealSolidSolnPhase::intEnergy_mole(), IdealGasPhase::intEnergy_mole(), IdealGasPhase::logStandardConc(), IdealGasPhase::pressure(), ThermoPhase::report(), PureFluidPhase::report(), MolalityVPSSTP::report(), HMWSoln::s_updatePitzer_CoeffWRTemp(), HMWSoln::s_updatePitzer_dlnMolalityActCoeff_dP(), HMWSoln::s_updatePitzer_lnMolalityActCoeff(), WaterSSTP::satPressure(), HMWSoln::satPressure(), Phase::saveState(), WaterSSTP::setDensity(), ThermoPhase::setElementPotentials(), WaterSSTP::setPressure(), VPStandardStateTP::setPressure(), IdealMolalSoln::setPressure(), IdealGasPhase::setPressure(), DebyeHuckel::setPressure(), SingleSpeciesTP::setState_HP(), ThermoPhase::setState_HPorUV(), SingleSpeciesTP::setState_SP(), ThermoPhase::setState_SPorSV(), SingleSpeciesTP::setState_SV(), SingleSpeciesTP::setState_UV(), IdealSolnGasVPSS::standardConcentration(), IdealGasPhase::standardConcentration(), MetalSHEelectrons::thermalExpansionCoeff(), IdealGasPhase::thermalExpansionCoeff(), VPStandardStateTP::updateStandardStateThermo(), and WaterSSTP::vaporFraction().
| doublereal thermalExpansionCoeff | ( | ) | const [virtual] |
The thermal expansion coefficient. Units: 1/K.
The thermal expansion coefficient is defined as
![\[ \beta = \frac{1}{v}\left(\frac{\partial v}{\partial T}\right)_P \]](form_96.png)
It's equal to zero for this model, since the molar volume doesn't change with pressure or temperature.
Reimplemented from ThermoPhase.
Definition at line 332 of file IdealMolalSoln.cpp.
| virtual void updateDensity | ( | ) | [inline, virtual, inherited] |
| void updateStandardStateThermo | ( | ) | const [virtual, inherited] |
Updates the standard state thermodynamic functions at the current T and P of the solution.
If m_useTmpStandardStateStorage is true, this function must be called for every call to functions in this class. It checks to see whether the temperature or pressure has changed and thus the ss thermodynamics functions for all of the species must be recalculated.
This function is responsible for updating the following internal members, when m_useTmpStandardStateStorage is true.
- m_hss_RT;
- m_cpss_R;
- m_gss_RT;
- m_sss_R;
- m_Vss
If m_useTmpStandardStateStorage is not true, this function may be required to be called by child classes to update internal member data.
Definition at line 501 of file VPStandardStateTP.cpp.
References VPStandardStateTP::_updateStandardStateThermo(), VPStandardStateTP::m_Pcurrent, VPStandardStateTP::m_Plast_ss, VPStandardStateTP::m_Tlast_ss, and State::temperature().
Referenced by IdealSolnGasVPSS::cp_mole(), IdealSolnGasVPSS::enthalpy_mole(), IdealSolnGasVPSS::entropy_mole(), HMWSoln::getActivities(), VPStandardStateTP::getCp_R(), VPStandardStateTP::getCp_R_ref(), VPStandardStateTP::getEnthalpy_RT(), VPStandardStateTP::getEnthalpy_RT_ref(), VPStandardStateTP::getEntropy_R(), VPStandardStateTP::getEntropy_R_ref(), VPStandardStateTP::getGibbs_ref(), VPStandardStateTP::getGibbs_RT(), VPStandardStateTP::getGibbs_RT_ref(), VPStandardStateTP::getIntEnergy_RT(), VPStandardStateTP::getPureGibbs(), VPStandardStateTP::getStandardVolumes(), VPStandardStateTP::getStandardVolumes_ref(), HMWSoln::getUnscaledMolalityActivityCoefficients(), VPStandardStateTP::setPressure(), IdealSolnGasVPSS::setPressure(), VPStandardStateTP::setState_TP(), IdealMolalSoln::setState_TP(), VPStandardStateTP::setTemperature(), MolalityVPSSTP::setToEquilState(), IdealSolnGasVPSS::setToEquilState(), and HMWSoln::setToEquilState().
| virtual doublereal vaporFraction | ( | ) | const [inline, virtual, inherited] |
Return the fraction of vapor at the current conditions.
Reimplemented in DebyeHuckel, HMWSoln, PureFluidPhase, SingleSpeciesTP, and WaterSSTP.
Definition at line 1822 of file ThermoPhase.h.
References ThermoPhase::err().
| XML_Node & xml | ( | ) | [inherited] |
Returns a reference to the XML_Node storred for the phase.
The XML_Node for the phase contains all of the input data used to set up the model for the phase, during its initialization.
Definition at line 112 of file Phase.cpp.
References Phase::m_xml.
Referenced by WaterSSTP::constructPhaseFile(), IdealSolidSolnPhase::constructPhaseFile(), IdealMolalSoln::constructPhaseFile(), HMWSoln::constructPhaseFile(), DebyeHuckel::constructPhaseFile(), and ThermoPhase::initThermoFile().
Member Data Documentation
| doublereal IMS_afCut_ |
Parameter in the polyExp cutoff treatment having to do with rate of exp decay.
Definition at line 974 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| doublereal IMS_agCut_ |
Parameter in the polyExp cutoff treatment having to do with rate of exp decay.
Definition at line 993 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| doublereal IMS_bfCut_ |
Parameter in the polyExp cutoff treatment having to do with rate of exp decay.
Definition at line 977 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| doublereal IMS_bgCut_ |
Parameter in the polyExp cutoff treatment having to do with rate of exp decay.
Definition at line 996 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| doublereal IMS_cCut_ |
Parameter in the polyExp cutoff treatment having to do with rate of exp decay.
Definition at line 958 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::initThermoXML(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| doublereal IMS_dfCut_ |
Parameter in the polyExp cutoff treatment having to do with rate of exp decay.
Definition at line 968 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| doublereal IMS_dgCut_ |
Parameter in the polyExp cutoff treatment having to do with rate of exp decay.
Definition at line 987 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| doublereal IMS_efCut_ |
Parameter in the polyExp cutoff treatment having to do with rate of exp decay.
Definition at line 971 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| doublereal IMS_egCut_ |
Parameter in the polyExp cutoff treatment having to do with rate of exp decay.
Definition at line 990 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| doublereal IMS_gamma_k_min_ |
gamma_k minimun for the cutoff process at the zero solvent point
Definition at line 955 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::initThermoXML(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| doublereal IMS_gamma_o_min_ |
gamma_o value for the cutoff process at the zero solvent point
Definition at line 952 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::initThermoXML(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
vector_fp IMS_lnActCoeffMolal_ [mutable, private] |
Logarithm of the molal activity coefficients.
Normally these are all one. However, stability schemes will change that
Definition at line 945 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::getActivities(), IdealMolalSoln::getChemPotentials(), IdealMolalSoln::getMolalityActivityCoefficients(), IdealMolalSoln::getPartialMolarEntropies(), IdealMolalSoln::initLengths(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| doublereal IMS_slopefCut_ |
Parameter in the polyExp cutoff treatment.
This is the slope of the f function at the zero solvent point Default value is 0.6
Definition at line 965 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::initThermoXML(), and IdealMolalSoln::operator=().
| doublereal IMS_slopegCut_ |
Parameter in the polyExp cutoff treatment.
This is the slope of the g function at the zero solvent point Default value is 0.0
Definition at line 984 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::initThermoXML(), and IdealMolalSoln::operator=().
| int IMS_typeCutoff_ |
Cutoff type.
Definition at line 916 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::getActivities(), IdealMolalSoln::getChemPotentials(), IdealMolalSoln::getMolalityActivityCoefficients(), IdealMolalSoln::getPartialMolarEntropies(), IdealMolalSoln::initThermoXML(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
| doublereal IMS_X_o_cutoff_ |
value of the solute mole fraction that centers the cutoff polynomials for the cutoff =1 process;
Definition at line 949 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcIMSCutoffParams_(), IdealMolalSoln::getChemPotentials(), IdealMolalSoln::initThermoXML(), IdealMolalSoln::operator=(), and IdealMolalSoln::s_updateIMS_lnMolalityActCoeff().
bool m_chargeNeutralityNecessary [protected, inherited] |
Boolean indicating whether a charge neutrality condition is a necessity.
Note, the charge neutrality condition is not a necessity for ideal gas phases. There may be a net charge in those phases, because the NASA polynomials for ionized species in Ideal gases take this condition into account. However, liquid phases usually require charge neutrality in order for their derived thermodynamics to be valid.
Definition at line 2130 of file ThermoPhase.h.
Referenced by ThermoPhase::chargeNeutralityNecessary(), MolalityVPSSTP::MolalityVPSSTP(), and ThermoPhase::operator=().
Elements* m_Elements [protected, inherited] |
Pointer to the element object corresponding to this phase. Normally, this will be the default Element object common to all phases.
Definition at line 366 of file Constituents.h.
Referenced by Constituents::addElement(), Constituents::addElementsFromXML(), Constituents::addUniqueElement(), Constituents::addUniqueSpecies(), Constituents::atomicNumber(), Constituents::atomicWeight(), Constituents::atomicWeights(), Constituents::Constituents(), Constituents::elementIndex(), Constituents::elementName(), Constituents::elementNames(), Constituents::elementsFrozen(), Constituents::entropyElement298(), Constituents::freezeElements(), Constituents::getAtoms(), Constituents::nAtoms(), Constituents::nElements(), Constituents::operator=(), Constituents::ready(), and Constituents::~Constituents().
vector_fp m_expg0_RT [mutable, private] |
Vector containing the species reference exp(-G/RT) functions at T = m_tlast.
Definition at line 924 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::initLengths(), and IdealMolalSoln::operator=().
int m_formGC [protected] |
The standard concentrations can have three different forms depending on the value of the member attribute m_formGC, which is supplied in the XML file.
| m_formGC | ActivityConc | StandardConc |
| 0 | | |
| 1 | | |
| 2 | | |
Definition at line 912 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::getActivityConcentrations(), IdealMolalSoln::initThermoXML(), IdealMolalSoln::operator=(), and IdealMolalSoln::standardConcentration().
bool m_hasElementPotentials [protected, inherited] |
Boolean indicating whether there is a valid set of saved element potentials for this phase.
Definition at line 2120 of file ThermoPhase.h.
Referenced by ThermoPhase::getElementPotentials(), ThermoPhase::operator=(), and ThermoPhase::setElementPotentials().
int m_index [protected, inherited] |
Index number of the phase.
The Cantera interface library uses this member to set the index number to the location of the pointer to this object in the pointer array of ThermoPhase's it maintains. Using this member for any other purpose will lead to unpredictable results if used in conjunction with the interface library.
Reimplemented from Phase.
Definition at line 2106 of file ThermoPhase.h.
Referenced by ThermoPhase::index(), ThermoPhase::operator=(), and ThermoPhase::setIndex().
int m_indexCLM [protected, inherited] |
Index of the phScale species.
Index of the species to be used in the single-ion scaling law. This is the indentity of the Cl- species for the PHSCALE_NBS scaling
Definition at line 872 of file MolalityVPSSTP.h.
Referenced by HMWSoln::applyphScale(), MolalityVPSSTP::initThermo(), MolalityVPSSTP::operator=(), HMWSoln::s_updateScaling_pHScaling(), HMWSoln::s_updateScaling_pHScaling_dT(), and HMWSoln::s_updateScaling_pHScaling_dT2().
int m_indexSolvent [protected, inherited] |
Index of the solvent.
Currently the index of the solvent is hard-coded to the value 0
Definition at line 854 of file MolalityVPSSTP.h.
Referenced by DebyeHuckel::_lnactivityWaterHelgesonFixedForm(), MolalityVPSSTP::calcMolalities(), IdealMolalSoln::getActivities(), HMWSoln::getActivities(), DebyeHuckel::getActivities(), MolalityVPSSTP::getActivityCoefficients(), IdealMolalSoln::getChemPotentials(), HMWSoln::getChemPotentials(), DebyeHuckel::getChemPotentials(), IdealMolalSoln::getMolalityActivityCoefficients(), IdealMolalSoln::getPartialMolarEntropies(), HMWSoln::getPartialMolarEntropies(), DebyeHuckel::getPartialMolarEntropies(), IdealMolalSoln::initThermoXML(), HMWSoln::initThermoXML(), DebyeHuckel::initThermoXML(), MolalityVPSSTP::operator=(), MolalityVPSSTP::osmoticCoefficient(), DebyeHuckel::s_update_d2lnMolalityActCoeff_dT2(), DebyeHuckel::s_update_dlnMolalityActCoeff_dP(), DebyeHuckel::s_update_dlnMolalityActCoeff_dT(), DebyeHuckel::s_update_lnMolalityActCoeff(), IdealMolalSoln::s_updateIMS_lnMolalityActCoeff(), HMWSoln::s_updateIMS_lnMolalityActCoeff(), HMWSoln::s_updatePitzer_d2lnMolalityActCoeff_dT2(), HMWSoln::s_updatePitzer_dlnMolalityActCoeff_dP(), HMWSoln::s_updatePitzer_dlnMolalityActCoeff_dT(), HMWSoln::s_updatePitzer_lnMolalityActCoeff(), MolalityVPSSTP::setMolalities(), MolalityVPSSTP::setMolalitiesByName(), MolalityVPSSTP::setSolvent(), MolalityVPSSTP::solventIndex(), IdealMolalSoln::standardConcentration(), HMWSoln::standardConcentration(), and DebyeHuckel::standardConcentration().
int m_kk [protected, inherited] |
m_kk = Number of species in the phase.
For internal use only.
m_kk is a member of both the State and Constituents classes. Therefore, to avoid multiple inheritance problems, we need to restate it in here, so that the declarations in the two base classes become hidden.
Reimplemented from Constituents.
Definition at line 504 of file Phase.h.
Referenced by DebyeHuckel::_lnactivityWaterHelgesonFixedForm(), SurfPhase::_updateThermo(), IdealSolidSolnPhase::_updateThermo(), IdealGasPhase::_updateThermo(), IdealMolalSoln::calcDensity(), DebyeHuckel::calcDensity(), MolalityVPSSTP::calcMolalities(), ConstDensityThermo::expGibbs_RT(), IdealSolidSolnPhase::expGibbs_RT_ref(), IdealGasPhase::expGibbs_RT_ref(), MolalityVPSSTP::findCLMIndex(), Phase::freezeSpecies(), IdealMolalSoln::getActivities(), DebyeHuckel::getActivities(), ThermoPhase::getActivityCoefficients(), SingleSpeciesTP::getActivityCoefficients(), MolalityVPSSTP::getActivityCoefficients(), IdealSolnGasVPSS::getActivityCoefficients(), IdealSolidSolnPhase::getActivityCoefficients(), IdealGasPhase::getActivityCoefficients(), IdealSolnGasVPSS::getActivityConcentrations(), IdealSolidSolnPhase::getActivityConcentrations(), IdealMolalSoln::getActivityConcentrations(), DebyeHuckel::getActivityConcentrations(), SurfPhase::getChemPotentials(), IdealSolnGasVPSS::getChemPotentials(), IdealSolidSolnPhase::getChemPotentials(), IdealMolalSoln::getChemPotentials(), IdealGasPhase::getChemPotentials(), DebyeHuckel::getChemPotentials(), VPStandardStateTP::getChemPotentials_RT(), IdealSolnGasVPSS::getChemPotentials_RT(), IdealSolidSolnPhase::getChemPotentials_RT(), SurfPhase::getCoverages(), IdealSolidSolnPhase::getCp_R_ref(), ThermoPhase::getElectrochemPotentials(), MolalityVPSSTP::getElectrochemPotentials(), IdealSolidSolnPhase::getEnthalpy_RT(), IdealSolidSolnPhase::getEnthalpy_RT_ref(), IdealGasPhase::getEntropy_R(), IdealSolidSolnPhase::getEntropy_R_ref(), WaterSSTP::getGibbs_ref(), IdealSolidSolnPhase::getGibbs_ref(), IdealSolidSolnPhase::getGibbs_RT(), IdealGasPhase::getGibbs_RT(), IdealSolidSolnPhase::getGibbs_RT_ref(), IdealSolidSolnPhase::getIntEnergy_RT(), IdealGasPhase::getIntEnergy_RT(), IdealSolidSolnPhase::getIntEnergy_RT_ref(), IdealGasPhase::getIntEnergy_RT_ref(), MolalityVPSSTP::getMolalities(), IdealMolalSoln::getMolalityActivityCoefficients(), DebyeHuckel::getMolalityActivityCoefficients(), SurfPhase::getPartialMolarCp(), IdealSolnGasVPSS::getPartialMolarCp(), IdealSolidSolnPhase::getPartialMolarCp(), IdealMolalSoln::getPartialMolarCp(), DebyeHuckel::getPartialMolarCp(), SurfPhase::getPartialMolarEnthalpies(), IdealSolnGasVPSS::getPartialMolarEnthalpies(), IdealMolalSoln::getPartialMolarEnthalpies(), DebyeHuckel::getPartialMolarEnthalpies(), SurfPhase::getPartialMolarEntropies(), IdealSolnGasVPSS::getPartialMolarEntropies(), IdealSolidSolnPhase::getPartialMolarEntropies(), IdealMolalSoln::getPartialMolarEntropies(), IdealGasPhase::getPartialMolarEntropies(), DebyeHuckel::getPartialMolarEntropies(), IdealSolnGasVPSS::getPartialMolarIntEnergies(), IdealGasPhase::getPartialMolarIntEnergies(), IdealGasPhase::getPartialMolarVolumes(), DebyeHuckel::getPartialMolarVolumes(), IdealSolidSolnPhase::getPureGibbs(), IdealGasPhase::getPureGibbs(), ThermoPhase::getReferenceComposition(), VPStandardStateTP::getStandardChemPotentials(), IdealGasPhase::getStandardChemPotentials(), SurfPhase::getStandardVolumes(), IdealGasPhase::getStandardVolumes(), IdealGasPhase::getStandardVolumes_ref(), HMWSoln::HMWSoln(), VPStandardStateTP::initLengths(), MolalityVPSSTP::initLengths(), IdealSolnGasVPSS::initLengths(), IdealSolidSolnPhase::initLengths(), IdealMolalSoln::initLengths(), DebyeHuckel::initLengths(), VPStandardStateTP::initThermo(), ThermoPhase::initThermo(), SurfPhase::initThermo(), StoichSubstanceSSTP::initThermo(), SingleSpeciesTP::initThermo(), IdealGasPhase::initThermo(), VPStandardStateTP::initThermoXML(), IdealSolidSolnPhase::initThermoXML(), IdealMolalSoln::initThermoXML(), HMWSoln::initThermoXML(), DebyeHuckel::initThermoXML(), IdealSolidSolnPhase::logStandardConc(), VPStandardStateTP::operator=(), ThermoPhase::operator=(), Phase::operator=(), MolalityVPSSTP::osmoticCoefficient(), HMWSoln::readXMLBinarySalt(), HMWSoln::readXMLLambdaNeutral(), HMWSoln::readXMLPsiCommonAnion(), HMWSoln::readXMLPsiCommonCation(), HMWSoln::readXMLThetaAnion(), HMWSoln::readXMLThetaCation(), HMWSoln::readXMLZetaCation(), Phase::ready(), IdealSolidSolnPhase::referenceConcentration(), DebyeHuckel::s_update_d2lnMolalityActCoeff_dT2(), DebyeHuckel::s_update_dlnMolalityActCoeff_dP(), DebyeHuckel::s_update_dlnMolalityActCoeff_dT(), DebyeHuckel::s_update_lnMolalityActCoeff(), IdealMolalSoln::s_updateIMS_lnMolalityActCoeff(), SurfPhase::setCoverages(), SurfPhase::setCoveragesNoNorm(), MolalityVPSSTP::setMolalities(), ThermoPhase::setReferenceComposition(), MolalityVPSSTP::setSolvent(), IdealSolnGasVPSS::setToEquilState(), IdealSolidSolnPhase::setToEquilState(), IdealGasPhase::setToEquilState(), ThermoPhase::speciesData(), IdealSolidSolnPhase::standardConcentration(), and ThermoPhase::~ThermoPhase().
vector_fp m_lambdaRRT [protected, inherited] |
Vector of element potentials.
-> length equal to number of elements
Definition at line 2116 of file ThermoPhase.h.
Referenced by ThermoPhase::getElementPotentials(), ThermoPhase::operator=(), and ThermoPhase::setElementPotentials().
doublereal m_Mnaught [protected, inherited] |
This is the multiplication factor that goes inside log expressions involving the molalities of species.
Its equal to Wt_0 / 1000, where Wt_0 = weight of solvent (kg/kmol)
Definition at line 893 of file MolalityVPSSTP.h.
Referenced by DebyeHuckel::_lnactivityWaterHelgesonFixedForm(), MolalityVPSSTP::calcMolalities(), MolalityVPSSTP::operator=(), MolalityVPSSTP::osmoticCoefficient(), DebyeHuckel::s_update_d2lnMolalityActCoeff_dT2(), DebyeHuckel::s_update_dlnMolalityActCoeff_dP(), DebyeHuckel::s_update_dlnMolalityActCoeff_dT(), DebyeHuckel::s_update_lnMolalityActCoeff(), MolalityVPSSTP::setMolalities(), MolalityVPSSTP::setMolalitiesByName(), MolalityVPSSTP::setSolvent(), and HMWSoln::standardConcentration().
vector_fp m_molalities [mutable, protected, inherited] |
Current value of the molalities of the species in the phase.
Note this vector is a mutable quantity. units are (kg/kmol)
Definition at line 900 of file MolalityVPSSTP.h.
Referenced by DebyeHuckel::_lnactivityWaterHelgesonFixedForm(), MolalityVPSSTP::calcMolalities(), IdealMolalSoln::getActivities(), HMWSoln::getActivities(), DebyeHuckel::getActivities(), IdealMolalSoln::getChemPotentials(), HMWSoln::getChemPotentials(), DebyeHuckel::getChemPotentials(), MolalityVPSSTP::getMolalities(), IdealMolalSoln::getPartialMolarEntropies(), HMWSoln::getPartialMolarEntropies(), DebyeHuckel::getPartialMolarEntropies(), MolalityVPSSTP::initLengths(), MolalityVPSSTP::operator=(), MolalityVPSSTP::osmoticCoefficient(), DebyeHuckel::s_update_d2lnMolalityActCoeff_dT2(), DebyeHuckel::s_update_dlnMolalityActCoeff_dP(), DebyeHuckel::s_update_dlnMolalityActCoeff_dT(), DebyeHuckel::s_update_lnMolalityActCoeff(), HMWSoln::s_updatePitzer_lnMolalityActCoeff(), and MolalityVPSSTP::setMolalities().
int m_ndim [protected, inherited] |
m_ndim is the dimensionality of the phase.
Volumetric phases have dimensionality 3 and surface phases have dimensionality 2.
Definition at line 511 of file Phase.h.
Referenced by Phase::nDim(), Phase::operator=(), and Phase::setNDim().
doublereal m_P0 [protected, inherited] |
Reference pressure (Pa) must be the same for all species
- defaults to OneAtm
Definition at line 624 of file VPStandardStateTP.h.
Referenced by VPStandardStateTP::operator=().
doublereal m_Pcurrent [protected, inherited] |
Current value of the pressure - state variable.
Because we are now using the pressure as a state variable, we need to carry it along within this object
units = Pascals
Definition at line 611 of file VPStandardStateTP.h.
Referenced by VPStandardStateTP::_updateStandardStateThermo(), IdealSolnGasVPSS::calcDensity(), IdealSolnGasVPSS::isothermalCompressibility(), VPStandardStateTP::operator=(), VPStandardStateTP::pressure(), HMWSoln::pressure(), DebyeHuckel::pressure(), IdealSolnGasVPSS::setPressure(), VPStandardStateTP::setState_TP(), IdealMolalSoln::setState_TP(), DebyeHuckel::setState_TP(), VPStandardStateTP::setTemperature(), DebyeHuckel::setTemperature(), and VPStandardStateTP::updateStandardStateThermo().
std::vector<PDSS *> m_PDSS_storage [protected, inherited] |
Storage for the PDSS objects for the species.
Storage is in species index order. VPStandardStateTp owns each of the objects. Copy operations are deep.
Definition at line 639 of file VPStandardStateTP.h.
Referenced by VPStandardStateTP::initThermo(), VPStandardStateTP::initThermoXML(), VPStandardStateTP::operator=(), and VPStandardStateTP::~VPStandardStateTP().
vector_fp m_pe [mutable, private] |
Vector of potential energies for the species.
Definition at line 929 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::initLengths(), and IdealMolalSoln::operator=().
doublereal m_phi [protected, inherited] |
Storred value of the electric potential for this phase.
Units are Volts
Definition at line 2112 of file ThermoPhase.h.
Referenced by ThermoPhase::electricPotential(), IdealMolalSoln::electricPotential(), ThermoPhase::operator=(), ThermoPhase::setElectricPotential(), and IdealMolalSoln::setElectricPotential().
int m_pHScalingType [protected, inherited] |
Scaling to be used for output of single-ion species activity coefficients.
Index of the species to be used in the single-ion scaling law. This is the indentity of the Cl- species for the PHSCALE_NBS scaling. Either PHSCALE_PITZER or PHSCALE_NBS
Definition at line 864 of file MolalityVPSSTP.h.
Referenced by HMWSoln::applyphScale(), MolalityVPSSTP::operator=(), MolalityVPSSTP::pHScale(), HMWSoln::s_updateScaling_pHScaling(), HMWSoln::s_updateScaling_pHScaling_dT(), HMWSoln::s_updateScaling_pHScaling_dT2(), and MolalityVPSSTP::setpHScale().
doublereal m_Plast_ss [mutable, protected, inherited] |
The last pressure at which the Standard State thermodynamic properties were calculated at.
Definition at line 618 of file VPStandardStateTP.h.
Referenced by VPStandardStateTP::_updateStandardStateThermo(), VPStandardStateTP::operator=(), and VPStandardStateTP::updateStandardStateThermo().
vector_fp m_pp [mutable, private] |
Temporary array used in equilibrium calculations.
Definition at line 934 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcDensity(), IdealMolalSoln::enthalpy_mole(), IdealMolalSoln::initLengths(), and IdealMolalSoln::operator=().
vector_fp m_speciesCharge [protected, inherited] |
m_speciesCharge: Vector of species charges length = m_kk
Definition at line 383 of file Constituents.h.
Referenced by Constituents::addUniqueSpecies(), HMWSoln::applyphScale(), Constituents::charge(), HMWSoln::initThermoXML(), DebyeHuckel::initThermoXML(), Constituents::operator=(), HMWSoln::readXMLBinarySalt(), HMWSoln::readXMLLambdaNeutral(), HMWSoln::readXMLMunnnNeutral(), HMWSoln::readXMLPsiCommonAnion(), HMWSoln::readXMLPsiCommonCation(), HMWSoln::readXMLThetaAnion(), HMWSoln::readXMLThetaCation(), HMWSoln::readXMLZetaCation(), DebyeHuckel::s_update_d2lnMolalityActCoeff_dT2(), DebyeHuckel::s_update_dlnMolalityActCoeff_dP(), DebyeHuckel::s_update_dlnMolalityActCoeff_dT(), DebyeHuckel::s_update_lnMolalityActCoeff(), HMWSoln::s_updatePitzer_CoeffWRTemp(), HMWSoln::s_updatePitzer_d2lnMolalityActCoeff_dT2(), HMWSoln::s_updatePitzer_dlnMolalityActCoeff_dP(), HMWSoln::s_updatePitzer_dlnMolalityActCoeff_dT(), HMWSoln::s_updatePitzer_lnMolalityActCoeff(), HMWSoln::s_updateScaling_pHScaling(), HMWSoln::s_updateScaling_pHScaling_dT(), and HMWSoln::s_updateScaling_pHScaling_dT2().
vector_fp m_speciesComp [protected, inherited] |
Atomic composition of the species.
the number of atoms of i in species k is equal to m_speciesComp[k * m_mm + i] The length of this vector is equal to m_kk * m_mm
Definition at line 377 of file Constituents.h.
Referenced by Constituents::addUniqueSpecies(), Constituents::getAtoms(), Constituents::nAtoms(), and Constituents::operator=().
std::vector<const XML_Node *> m_speciesData [protected, inherited] |
Vector of pointers to the species databases.
This is used to access data needed to construct the transport manager and other properties later in the initialization process. We create a copy of the XML_Node data read in here. Therefore, we own this data.
Definition at line 2096 of file ThermoPhase.h.
Referenced by ThermoPhase::operator=(), ThermoPhase::saveSpeciesData(), ThermoPhase::speciesData(), and ThermoPhase::~ThermoPhase().
bool m_speciesFrozen [protected, inherited] |
Boolean indicating whether the number of species has been frozen.
During the construction of the phase, this is false. After construction of the the phase, this is true.
Definition at line 359 of file Constituents.h.
Referenced by Constituents::freezeSpecies(), Constituents::operator=(), Constituents::ready(), and Constituents::speciesFrozen().
array_fp m_speciesMolarVolume [protected] |
Species molar volume .
Definition at line 898 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::initLengths(), IdealMolalSoln::initThermoXML(), IdealMolalSoln::operator=(), and IdealMolalSoln::standardConcentration().
std::vector<std::string> m_speciesNames [protected, inherited] |
Vector of the species names.
Definition at line 369 of file Constituents.h.
Referenced by Constituents::addUniqueSpecies(), Constituents::operator=(), Constituents::speciesIndex(), Constituents::speciesName(), and Constituents::speciesNames().
vector_fp m_speciesSize [protected, inherited] |
m_speciesSize(): Vector of species sizes.
length m_kk This is used in some equations of state which employ the constant partial molar volume approximation. It's so fundamental we've put it at the Constituents class level
Definition at line 393 of file Constituents.h.
Referenced by Constituents::addUniqueSpecies(), HMWSoln::initLengths(), DebyeHuckel::initLengths(), MineralEQ3::initThermoXML(), HMWSoln::initThermoXML(), DebyeHuckel::initThermoXML(), Constituents::operator=(), Constituents::size(), and DebyeHuckel::standardConcentration().
SpeciesThermo* m_spthermo [protected, inherited] |
Pointer to the calculation manager for species reference-state thermodynamic properties.
This class is called when the reference-state thermodynamic properties of all the species in the phase needs to be evaluated.
Definition at line 2086 of file ThermoPhase.h.
Referenced by SurfPhase::_updateThermo(), SingleSpeciesTP::_updateThermo(), IdealSolidSolnPhase::_updateThermo(), IdealGasPhase::_updateThermo(), IdealGasPhase::entropy_mole(), IdealGasPhase::getEntropy_R(), IdealGasPhase::getGibbs_RT(), IdealGasPhase::getPartialMolarEntropies(), IdealGasPhase::getPureGibbs(), IdealGasPhase::getStandardChemPotentials(), IdealSolidSolnPhase::initLengths(), StoichSubstanceSSTP::initThermo(), SingleSpeciesTP::initThermo(), IdealGasPhase::initThermo(), WaterSSTP::initThermoXML(), ThermoPhase::maxTemp(), ThermoPhase::minTemp(), VPStandardStateTP::operator=(), ThermoPhase::operator=(), ThermoPhase::refPressure(), ThermoPhase::setSpeciesThermo(), ThermoPhase::speciesThermo(), IdealMolalSoln::speciesThermo(), HMWSoln::speciesThermo(), DebyeHuckel::speciesThermo(), and ThermoPhase::~ThermoPhase().
int m_ssConvention [protected, inherited] |
Contains the standard state convention.
Definition at line 2133 of file ThermoPhase.h.
Referenced by ThermoPhase::operator=(), and ThermoPhase::standardStateConvention().
doublereal m_Tlast_ss [mutable, protected, inherited] |
The last temperature at which the standard statethermodynamic properties were calculated at.
Definition at line 614 of file VPStandardStateTP.h.
Referenced by VPStandardStateTP::_updateStandardStateThermo(), VPStandardStateTP::operator=(), and VPStandardStateTP::updateStandardStateThermo().
vector_fp m_tmpV [mutable, private] |
vector of size m_kk, used as a temporary holding area.
Definition at line 939 of file IdealMolalSoln.h.
Referenced by IdealMolalSoln::calcDensity(), IdealMolalSoln::cp_mole(), IdealMolalSoln::enthalpy_mole(), IdealMolalSoln::entropy_mole(), IdealMolalSoln::gibbs_mole(), IdealMolalSoln::initLengths(), IdealMolalSoln::intEnergy_mole(), and IdealMolalSoln::operator=().
VPSSMgr* m_VPSS_ptr [mutable, protected, inherited] |
Pointer to the VPSS manager that calculates all of the standard state info efficiently.
Definition at line 631 of file VPStandardStateTP.h.
Referenced by VPStandardStateTP::_updateStandardStateThermo(), IdealSolnGasVPSS::calcDensity(), IdealSolnGasVPSS::cp_mole(), IdealSolnGasVPSS::enthalpy_mole(), IdealSolnGasVPSS::entropy_mole(), IdealSolnGasVPSS::getActivityConcentrations(), VPStandardStateTP::getCp_R(), VPStandardStateTP::getCp_R_ref(), VPStandardStateTP::getEnthalpy_RT(), VPStandardStateTP::getEnthalpy_RT_ref(), VPStandardStateTP::getEntropy_R(), VPStandardStateTP::getEntropy_R_ref(), VPStandardStateTP::getGibbs_ref(), VPStandardStateTP::getGibbs_RT(), VPStandardStateTP::getGibbs_RT_ref(), VPStandardStateTP::getIntEnergy_RT(), VPStandardStateTP::getPureGibbs(), VPStandardStateTP::getStandardVolumes(), VPStandardStateTP::getStandardVolumes_ref(), VPStandardStateTP::initThermo(), VPStandardStateTP::initThermoXML(), VPStandardStateTP::operator=(), VPStandardStateTP::provideVPSSMgr(), IdealSolnGasVPSS::setToEquilState(), VPStandardStateTP::setVPSSMgr(), IdealSolnGasVPSS::standardConcentration(), and VPStandardStateTP::~VPStandardStateTP().
vector_fp m_weight [protected, inherited] |
Vector of molecular weights of the species.
This vector has length m_kk. The units of the vector are kg kmol-1.
Definition at line 352 of file Constituents.h.
Referenced by WaterSSTP::initThermoXML(), Constituents::molecularWeight(), Constituents::molecularWeights(), and Constituents::operator=().
doublereal m_weightSolvent [protected, inherited] |
Molecular weight of the Solvent.
Definition at line 875 of file MolalityVPSSTP.h.
Referenced by MolalityVPSSTP::operator=(), HMWSoln::s_updatePitzer_d2lnMolalityActCoeff_dT2(), HMWSoln::s_updatePitzer_dlnMolalityActCoeff_dP(), HMWSoln::s_updatePitzer_dlnMolalityActCoeff_dT(), HMWSoln::s_updatePitzer_lnMolalityActCoeff(), and MolalityVPSSTP::setSolvent().
doublereal m_xmolSolventMIN [protected, inherited] |
In any molality implementation, it makes sense to have a minimum solvent mole fraction requirement, since the implementation becomes singular in the xmolSolvent=0 limit. The default is to set it to 0.01. We then modify the molality definition to ensure that molal_solvent = 0 when xmol_solvent = 0.
Definition at line 885 of file MolalityVPSSTP.h.
Referenced by MolalityVPSSTP::calcMolalities(), IdealMolalSoln::getActivities(), MolalityVPSSTP::getActivityCoefficients(), IdealMolalSoln::getMolalityActivityCoefficients(), MolalityVPSSTP::moleFSolventMin(), MolalityVPSSTP::operator=(), IdealMolalSoln::s_updateIMS_lnMolalityActCoeff(), HMWSoln::s_updateIMS_lnMolalityActCoeff(), HMWSoln::s_updatePitzer_lnMolalityActCoeff(), MolalityVPSSTP::setMolalitiesByName(), and MolalityVPSSTP::setMoleFSolventMin().
std::vector<doublereal> xMol_Ref [protected, inherited] |
Reference Mole Fraction Composition.
Occasionally, the need arises to find a safe mole fraction vector to initialize the object to. This contains such a vector. The algorithm will pick up the mole fraction vector that is applied from the state xml file in the input file
Definition at line 2142 of file ThermoPhase.h.
Referenced by ThermoPhase::getReferenceComposition(), ThermoPhase::initThermo(), and ThermoPhase::setReferenceComposition().
The documentation for this class was generated from the following files:
