
New computational chemistry techniques accelerate the prediction of molecules and materials
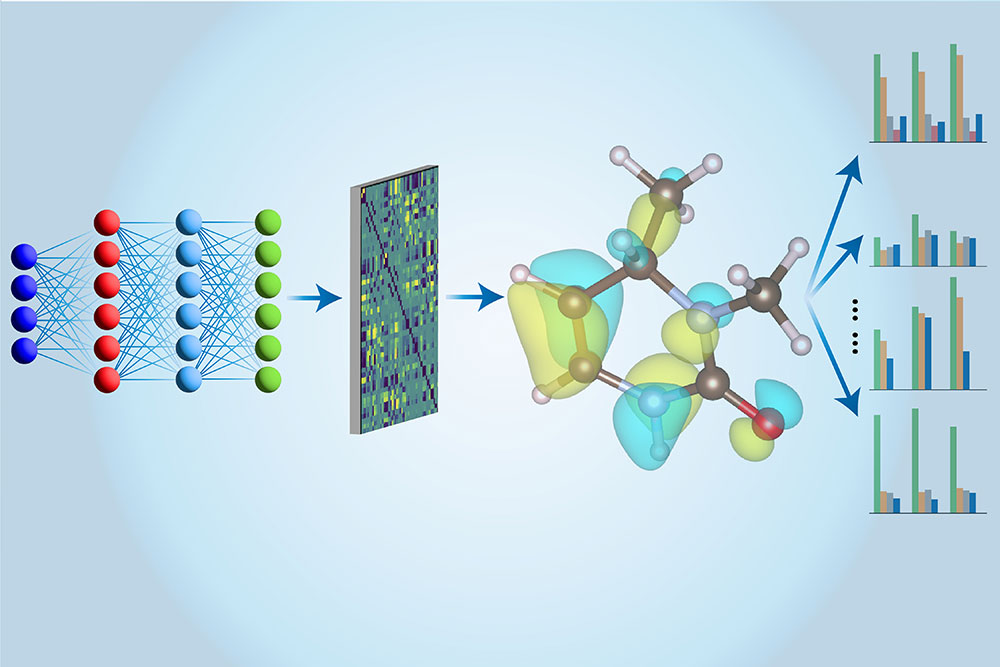
A multi-task machine learning approach is developed to predict the electronic properties of molecules, as demonstrated in the computational workflow illustrated here.
Back in the old days, the really old days, the task of designing materials was laborious. Investigators, over the course of 1000-plus years, tried to make gold by combining things like lead, mercury, and sulfur, mixed in what they hoped would be just the right proportions. Even famous scientists like Tycho Brahe, Robert Boyle, and Isaac Newton tried their hands at the fruitless endeavor we call alchemy.
Materials science has, of course, come a long way. For the past 150 years, researchers have had the benefit of the periodic table of elements to draw upon, which tells them that different elements have different properties, and one can’t magically transform into another. Moreover, in the past decade or so, machine learning tools have considerably boosted our capacity to determine the structure and physical properties of various molecules and substances. New research by a group led by Ju Li at MIT — the Tokyo Electric Power Company Professor of Nuclear Engineering and Professor of Materials Science and Engineering — offers the promise of a major leap in capabilities that can facilitate materials design. The results of their investigation are reported in January 2025 issue of Nature Computational Science.
At present, most of the machine learning models that are used to characterize molecular systems are based on density functional theory (DFT), which offers a quantum mechanical approach to determining the total energy of a molecule or crystal by looking at the electron density distribution, which is, basically, the average number of electrons located in a unit volume around each given point in space near the molecule. (Walter Kohn, who co-invented this theory 60 years ago, received a Nobel Prize in Chemistry in 1998.) While the method has been very successful, it has some drawbacks, according to Li. “First, the accuracy is not uniformly great. And, second, it only tells you one thing: the lowest total energy of the molecular system.”
“Couples therapy” to the rescue
His team is now relying on a different computational chemistry technique, also derived from quantum mechanics, known as coupled-cluster theory or CCSD(T). “This is the gold standard of quantum chemistry,” Li commented. The results of CCSD(T) calculations are much more accurate than what you get from DFT calculations, and they can be as trustworthy as those currently obtainable from experiments. The problem is that carrying out these calculations on a computer is very slow, he said, “and the scaling is bad: If you double the number of electrons in the system, the computations become 100 times more expensive.” For that reason, CCSD(T) calculations have normally been limited to molecules with a small number of atoms — on the order of about 10. Anything much beyond that would simply take too long.
That’s where machine learning comes in. CCSD(T) calculations are first performed on conventional computers, and the results are then used to train a neural network with a novel architecture specially devised by Li and his colleagues. After training, the neural network can perform these same calculations much faster by taking advantage of approximation techniques. What’s more, their neural network model can extract much more information about a molecule than just its energy. “In previous work, people have used multiple different models to assess different properties,” said Hao Tang, an MIT PhD student in Materials Science and Engineering. “Here we use just one model to evaluate all of these properties, which is why we call it a ‘multi-task’ approach.”
The “Multi-task Electronic Hamiltonian network”, or MEHnet, sheds light on a number of electronic properties, such as the dipole and quadrupole moments, electronic polarizability, and the optical excitation gap — the amount of energy needed to take an electron from the ground state to the lowest excited state. “The excitation gap affects the optical properties of materials,” Tang explained, “because it determines the frequency of light that can be absorbed by a molecule.” Another advantage of their CCSD-trained model is that it can reveal properties of not only ground states but also excited states. The model can also predict the infrared absorption spectrum of a molecule related to its vibrational properties, where the vibrations of atoms within a molecule are coupled to each other, leading to various collective behaviors.
The strength of their approach owes a lot to the network architecture. Drawing on the work of MIT Professor Tess Smidt, the team is utilizing a so-called E(3)-equivariant graph neural network said Tang, “in which the nodes represent atoms and the edges that connect the nodes represent the bonds between atoms. We also use customized algorithms that incorporate physics principles — related to how people calculate molecular properties in quantum mechanics — directly into our model.”
Testing, 1, 2 3
When tested on its analysis of known hydrocarbon molecules, the model of Li et al. outperformed DFT counterparts and closely matched experimental results taken from the published literature.
Qiang Zhu — a materials discovery specialist at the University of North Carolina, Charlotte (who was not part of this study) — is impressed by what’s been accomplished so far. “Their method enables effective training with a small dataset, while achieving superior accuracy and computational efficiency compared to existing models,” he said. “This is exciting work that illustrates the powerful synergy between computational chemistry and deep learning, offering fresh ideas for developing more accurate and scalable electronic structure methods.”
The MIT-based group applied their model first to small, non-metallic elements — hydrogen, carbon, nitrogen, oxygen, and fluorine from which organic compounds can be made — and has since moved on to examining heavier elements: silicon, phosphorus, sulfur, chlorine, and even platinum. After being trained on small molecules, the model can be generalized to bigger and bigger molecules. “Previously, most calculations were limited to analyzing hundreds of atoms with DFT and just tens of atoms with CCSD(T) calculations,” Li said. “Now we're talking about handling thousands of atoms and, eventually, perhaps tens of thousands.”
For now, the researchers are still evaluating known molecules, but the model can be used to characterize molecules that haven’t been seen before, as well as to predict the properties of hypothetical materials that consist of different kinds of molecules. “The idea is to use our theoretical tools to pick out promising candidates, which satisfy a particular set of criteria, before suggesting them to an experimentalist to check out,” Tang said.
It’s all about the apps
Looking ahead, Zhu is optimistic about the possible applications. “This approach holds the potential for high-throughput molecular screening,” he said. “That’s a task where achieving chemical accuracy can be essential for identifying novel molecules and materials with desirable properties.”
Once they demonstrate the ability to analyze large molecules with perhaps tens of thousands of atoms, Li said, “we should be able to invent new polymers or materials” that might be used in drug design or in semiconductor devices. The examination of heavier transition metal elements could lead to the advent of new materials for batteries – presently an area of acute need.
The future, as Li sees it, is wide open. “It’s no longer about just one area,” he said. “Our ambition, ultimately, is to cover the whole periodic table with CCSD(T)-level accuracy but at lower computational cost than DFT. This should enable us to solve a wide range of problems in chemistry, biology, and materials science. It’s hard to know, at present, just how wide that range might be.”
This work was supported by the Honda Research Institute (HRI-USA). Hao Tang acknowledges support from the Mathworks Engineering Fellowship. The calculations in this work were performed, in part, on the Matlantis high-speed universal atomistic simulator, the Texas Advanced Computing Center (TACC), the MIT SuperCloud, and the National Energy Research Scientific Computing (NERSC).
January 2025. Written by Steve Nadis. Image courtesy of the researchers.
Department of Nuclear Science & Engineering
Massachusetts Institute of Technology
77 Massachusetts Avenue, 24-107 (map)
Cambridge, MA 02139
nse-info@mit.edu